
Abstract
The male-specific region of Y chromosome (MSY) has accumulated a higher density of human endogenous retroviruses (HERVs) and related sequences when compared with other regions of the human genome. Here, we focused on one HERV family, HERV-K14C that seemed to integrate preferentially into the Y chromosome in humans. To identify every copies of HERV-K14C in the human genome, we applied computational screening to map precisely the locus of individual HERV-K14C copies. Interestingly, 29 of all 146 copies were located in Y chromosome, and these 29 copies were mostly dispersed in the palindromic region. Three distinct HERV-K14C-related transcripts were found and were exclusively expressed in human testis tissue. Based on our phylogenetic analysis of the solitary LTRs derived from HERV-K14C on the Y chromosome we suggested that these sequences were generated as pairs of identical sequences. Specifically, analysis of HERV-K14C-related sequences in the palindromic region demonstrated that the Y chromosomal amplicons existed in our common ancestors and the duplicated pairs arose after divergence of great apes approximately 8–10 million years ago. Taken together, our observation suggested that HERV-K14C-related sequences contributed to genomic diversification of Y chromosome during speciation of great ape lineage.
Similar content being viewed by others


Introduction
The human Y chromosome is believed to have evolved from an autosomal ancestor. The pseudoautosomal regions (PAR) at the termini of the X and Y chromosome recombine during meiosis, but most of parts of the X and Y chromosome do not undergo X-Y genetic exchanges. The male-specific region of Y chromosome (MSY) is directly inherited from father to son and is, therefore, highly informative regarding the evolutionary history of male lineages. The MSY contains at least 156 transcript units that seem to be expressed abundantly and exclusively in testis suggesting that these male-specific transcripts are associated with spermatogenesis.1
Roughly, 41 mega bases (Mb) of MSY is heterochomatic sequences, and most of the heterochromatin comprises the distal long arm of the Y chromosome. The euchromatic region of MSY, which spans 23 Mb, has three structurally distinct features, X-degenerated, X-transposed, and ampliconic sequences. The ampliconic region make up 25% of the euchromatic portion of the human MSY, which includes eight palindromes with pairs of duplicated amplicon that ranges from 9 Kb (kilo bases) to 1.45 Mb (Figure 1). There is approximately 99.97% sequence identity between the repeats within each palindrome, implying that the palindromes arose through duplication events approximately 10 million years ago.2 Based on sequence comparisons of the inner boundaries of each palindrome (P), six out of eight palindromes (P1, P2, P4, P6, P7, and P8) seem to have arisen after divergence of the great apes lineage suggesting that structures within the MSY has evolved recently. In particular, these palindromic sequences contained a high frequency (11.8%) of human endogenous retroviral sequences (HERVs) when compared to the average from the human whole genome (8%).3, 4, 5

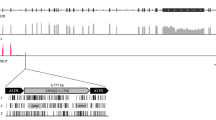
The overview of the structure of the Y chromosome. The male-specific region of Y chromosome (MSY). Black signifies the pseudoautosomal region, pale grey signifies euchromatin and dark grey is depicted as heterochromatic region of Y chromosome, respectively. Red triangles are depicted as locations of HERV-K14C locus on Table 2. (a) Enlarged schematic representation of the palindromic region, paralleled blue arrows represent each arm of eight palindromes. (b) Individual amplicons span from b1 on P3 to b4 on P1. Identical pairs of amplicons are colored the same color and arrows denote the direction of amplicons (c). The genomic structure of HERV-K14C-related transcripts (d). Exons are drawn as pale grey boxes and open reading frames are in black. The numbers above the boxes indicate exon number. Dark grey arrow depicts the HERV-K14C-related sequences, and the direction of the arrows illustrates the direction of transcription. Small black arrows indicate primer sequences (d). Vertical red arrows indicate the locations are used for genomic amplification in Figure 5 and represent a numbers in highlighted numbers on Table 2.
Several distinct families of HERVs exist in the human genome.6, 7 The majority of HERVs inserted into primate genome after the divergence of New World and Old World monkeys and were subjected to several amplification events during primate evolution.8, 9 When HERVs integrate into a host genome, they generate a form of the viral genome, including a 5′ LTR (long terminal repeats)—gag (capsid protein)—pol (viral enzyme)—env (envelope protein) -3′LTR, that is for replication and infection.10 Most HERVs are thought to be remnants of ancient germ line infection because they are defective and have accumulated multiple stop codons and deleterious mutations during evolution. Nevertheless, some HERV-related sequences are actively expressed and able to influence the host genome. For instance, the envelope protein of one of the most abundant families, HERV-W, serves an important function in the morphogenesis of the placenta in human.11 Not only the coding regions of HERVs contribute to transcriptome diversity; the LTRs contain transcription factor binding sites, hormone response elements, and polyadenylation signals that influence the host genome. Many reports demonstrate that HERV sequences regulate expression of functional genes near the site of integration.12, 13, 14
An ancient member of HERV-K family entered the genome in the Old World monkey lineage but HERV-K recurrently amplified and expanded during primate evolution.15 Some of HERV-K members are present only in chimpanzees and humans, indicating a relatively recent integration event within in the last five to eight million years.16 The solitary HERV-K LTR sequences, rather than the full-length retrovirus, give rise to the genetic variants found in some human individuals.17, 18 In addition, the amplification of HERV-K elements within the human lineage causes insertion polymorphism in the human genome.19
High sequence similarities between different HERV copies at different loci mediate non-reciprocal homologous recombination causing the accumulation of deleterious mutations in the human genome. For instance, recombination between HERV15 proviruses resulted in the deletion of the azoospermia factor a (AZFa) region on MSY and caused spermatogenic impairment.20 These previously published reports suggest that HERVs are a potential source of genetic diversity due not only to integration events but also to providing transcriptome variations.
The HERV-K14C element entered the primate germ line after the divergence between the Old World and New World monkey lineages approximately 39 million years ago.21 Flockerzi et al. surmised that 12 out of 23 copies of HERV-K14C copies were located on Y chromosome. Based on this knowledge, we hypothesized that the HERV-K14C seemed relevant to Y chromosomal evolution and we aimed to investigate this possibility with a high-resolution analysis of HERV-K14C elements on the Y chromosome. Indeed, our computational analysis demonstrates that copies of HERV-K14C were heavily dispersed in palindromic regions. We investigated a multilateral effect of HERV-K14C during Y chromosome evolution.
Materials and methods
Bioinformatic analysis
All human sequences were obtained from the NCBI database, Build 36. 1. HERV-K14C elements were identified in the Repbase database (http://www.girinst.org/repbase/) and in the genomic sequences using RepeatMasker (http://ftp.genome.washington.edu.cgi-bin/RepeatMasker), which uses a cross_match program to perform perfect sequence alignments.5, 22 As a consensus sequences, LTR14C and HERV-K14C consensus sequences were joined. Finally, LTR14C-HERV-K14C-LTR14C sequences were consensus sequences for our study. The internal retroviral sequences of the HERV-K14C were constructed by comparing conserved residues as potential coding region (gag, pro, pol, env) using the Blastx program.23 The human expressed sequence tag (EST), RefSeq mRNA, and non-redundant databases were screened using BLAST, version 2.2.11 with the ‘-q -e -e 0.01 –F T’ options, and HERV-K14C consensus sequences to identify hybrid transcripts.24 HERV-K14C-related transcripts (more than 95% sequence identity) were identified by comparing HERV-K14C elements with each database.
Phylogenetic analysis
Multiple alignment analysis and phylogenetic trees were constructed using loci containing solitary LTRs.25 The solitary LTRs from the human genome were selected to align with consensus sequence of LTR14C and these were matched over 80% of sequence identity. There were 93 loci identified in this study. To improve the accuracy of the comparisons, we limited our analysis to sequences greater or equal to roughly 500–600 bp. In addition, solitary LTRs were deleterious sequences or disturbed by insertions of other genomic sequences that 17 of 93 loci loci were eliminated. In all, 76 LTRs were analyzed to construct for phylogenetic tree. For a smooth phylogenetic analysis, incomplete and elements and elements containing many gaps were manually excluded. The neighbor-joining trees were obtained with the MEGA4 program.26 Bootstrap values for branches were calculated based on 1000 replications. Distances were estimated by the Kimura two-parameter method in the MEGA4 program to estimated sequence divergence.
Preparation of genomic and RNA samples
Total RNA from Japanese monkey tissues was extracted by RNeasy Mini Kit (Qiagen, Hilden, Germany), and total RNA from human tissue (brain, liver, lung, testis, heart, stomach, spleen, prostate, bladder, and ovary) was purchased from Clonetech (Clontech Laboratories, Inc, Mountain View, CA, USA). Pure mRNA was isolated using PolyA Tract mRNA isolation systems (Promega Corporation, Madison, WI, USA). Genomic DNA was isolated from blood samples by a standard protocol from the following species: (1) hominoids: chimpanzee (Pan troglodytes), gorilla (Gorilla gorilla), orangutan (Pongo pygmaeus), and gibbons (Hylobates agilis); (2) Old World monkeys: Japanese monkey (Macaca fuscata), rhesus monkey (Macaca mulatta); (3) New World monkeys: night monkey (Aotus trivirgatus), and common marmoset (Callithrix jacchus) using a standard protocol.
Genomic and RT-PCR amplification
cDNA samples from human and Japanese monkey RNA were to be synthesized by MMLV-derived reverse transcriptase with oligo (dT) and random hexamer primers (Promega Corporation). Genomic PCR and RT-PCR reactions were carried out using the standard PCR kit supplied by Takara. Primer information and accession numbers are documented in Table 1. SRY and GAPDH were used as positive controls.
Sequencing
PCR products amplified from genomic DNA were isolated using a PCR purification kit (Qiagen, Hilden, Germany) and were sequenced with the BigDye Terminator version 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) on an ABI wi337 DNA sequencer using the same primer pairs that had been used for amplification.
Statistical analysis
The difference between expected and observed copy number of HERV-K14C in proportions were contrasted by computing odds ratios (OR) and 95% CIs. All analyses were performed using Statistical Package for Social Sciences, version 11.0 (SPSS, Chicago, IL, USA) and the P-value was corrected using Fisher's exact test.
Results and Discussion
We first identified the genomic locations of all HERV-K14C-related sequences using computational approaches to search the entire human genome. In total, 146 loci dispersed across the human chromosomes. Only 14 of these loci had comparatively intact copies of HERV-K14C, and 93 loci had LTR-related sequences (Supplementary Table 1). The other 39 loci were deleted and mutated their internal sequences that mostly lost a 5′ or 3′ LTR. Presumably, the high frequency of solitary LTR copies implied that the majority of the HERV-K14C inserts underwent intra-homologous recombination between 5′ and 3′ LTR and then recurrently amplified in human genome. In addition, eight transcripts with HERV-K14C-related sequences were found suggesting that HERV-K14C elements provided promoters, exons, and polyadenylation signals for endogenous genes near the HERV insertion sites (Supplementary Table 2). Of these eight HERV-K-related transcripts, three resided in the MSY (Figure 1d).
The chromosomal distribution of HERV-K14C loci suggested that these sequences were not evenly dispersed in the human genome. HERV-K14C-related sequences were absent from chromosomes 9 and 22, whereas a high frequency of all HERV-K14C-related sequences (up to 20% or 29 copies) were found on the Y chromosome (Figure 2). To determine the statistical significance of high frequency of HERV-K14C copies on Y chromosome, we calculated the expected copy numbers based on the size of each chromosome. As shown in Figure 2, chromosome 8, 19 and Y seemed to present higher density of HERV-K14C copies when compared with expected copy numbers. Specifically, based on its size of Y chromosome was expected, theoretically, to have 3.09 copies of HERV-K14C element. However only Y chromosome had a statistical significance between expected and observed copy numbers (P<0.001, OR=11.8, 95% CI: 3.5–39.8). Indeed, HERV-K14C sequences were more likely to insert onto the Y chromosome than onto any of autosomes or the X chromosome.

The chromosomal distribution of HERV-K14C-related sequences. Pale grey bar depicts the expected copy number of HERV-K14C-related sequences based on the genomic size of each chromosome. Dark grey bar presents observed copy number of HERV-K14C within each chromosome. The P-value was calculated and corrected between expected copy number and observed copy number. The value of P>0.05 was not described.
To understand the evolutionary relevance of HERV-K14C sequences, we chose to study solitary LTRs for phylogenetic analysis because other fragment of HERV-K14C-related sequences were too truncated and mutated for useful analysis. Having eliminated 17 incomplete LTR copies, we used 76 copies of solitary LTR for this study (Supplementary Table 1). The neighbor-joining method was used to construct a phylogenetic tree with all 76 intact solitary LTR loci (Figure 3). Of the 76 LTR loci in the genome, 15 were located on the Y chromosome (Table 2). These 15 loci are marked with starred numbers in Figure 3, and the other loci are named by their chromosomal location.

The phylogenetic analysis of solitary LTRs from the entire genome. Bootstrap values derived from 1000 bootstrap replicates measured by the posterior probabilities are shown at the nodes. We also constructed a neighbor-joining tree using the MEGA4 program and the HERV-K14C consensus sequence. Each taxon name indicates the chromosomal location of a LTR locus except for those loci on the Y chromosome. Starred numbers detailed in Table 2 indicate the solitary LTRs on the Y chromosome.
As shown in Figure 3, the 76 solitary LTRs clustered into distinct phylogenetic groups that suggest these sequences were amplified at least four times after the initial genome integration event. Members of group I were scattered on autosome and both sex chromosomes. In contrast, no members of group II were found on the Y chromosome. One interesting features of this tree was that group I members derived from the Y chromosome clustered into same nodes. For instance, LTRs 7&14, 27&28, 9&12, and 10&11 formed five distinct pairs and LTRs 24, 25,26, and 29 clustered into a single node. These observations implied that each pair of LTRs was duplicated from a single copy of LTR during Y chromosomal evolutionary past. As we described above, HERV-K14C-related sequences were enriched on Y chromosome, and this observation explained, in part this feature of the phylogenetic analysis of the solitary LTRs. The insertion of initial HERV-K14C element predated to integrate into the Y chromosome, and HERV-K14C element underwent subsequent genomic amplification events producing identical pairs.
The copies of HERV-K14C-related sequences on the Y chromosome are listed in Table 2 and Figures 1a-c. As we mentioned above, 29 different HERV-K14C-related loci were identified on the Y chromosome; of these 17 loci were solitary LTRs and eight copies preserved both the 5′ and 3′ LTR. Three of loci only had a side of LTRs and its internal sequences; 5′LTR (8 and 13) and 3′LTR (3). In all, 18 copies were part of palindromes, meaning that these 18 copies actually represented 9 pairs, each with one member of the part on an arm of the respective palindrome. For example, 7–10 resided on the proximal arm of P5 and 11–14 resided on the distal arm of P5, the pairs were in reverse complementary direction. In a similar manner, two pairs—15/19 and 16/18 –were identical sequences in P4. The b amplicons had four of copies (24/25/26/29) scattered along P3 (b1 and b2) and P1 (b3 and b4) and these four duplicated copies were generated from a single HERV-K14C element. The duplicated pair (27/28) was found on P1.1 and P1.2. In each case, the sequence alignment between duplicated copies within a palindrome showed perfect identity between the duplicated pair. The solitary LTRs derived from the b amplicons were the only exceptions, there were single nucleotide difference (C → A) between the pairs 24/29 and 25/26. Four comparatively long HERV-K14C-related sequences—8, 13, 16, and 18 –also revealed perfect matched pairs (8&13 and 16&18) that did not have a single nucleotide gap or difference.
Although, 24, 25, 26 and 29 loci were all located in the b amplicons, they arose from different two duplication lineages. As shown in the topology of the tree of solitary LTRs on Y chromosome in Figure 4, duplicated partners clustered together. Each LTR copy on an arm of a palindrome was clearly clustered with one LTR copy on the other arm of the same palindrome. For example, copy 24 formed a pair with 29 and 25 formed a pair with 26 on the b amplicons. As illustrated in Figure 1c, P1 contains the b3/b4 amplicon as an inner segment and b1/b2 amplicon is dispersed across P3. According to our results, the 24 (b1)/29 (b4) and 25 (b2)/26 (b3) pairs were mingled indicating that genomic structures of P1 and P3 were generated in concert.

Fine scale phylogeny of the solitary LTRs on the Y chromosome. Only the gross topology of the tree is shown in this image. Bootstrap replicates support from 1000 is indicated for each node. Each taxon is described in Table 2.
To investigate, the integration history of each HERV-K14C copy on the Y chromosome, we looked for the orthologs of the boundaries each of HERV-K14C locus on Y chromosome to estimate an ancestral structure of the Y chromosome. Unlike autosomal chromosomes, the Y chromosome contains massive palindromes and many repeated sequences making it very difficult to amplify specific loci. Therefore, we chose fifteen HERV-K14C-related copies for amplification of genomic DNA from male primate genomes. The chosen loci are denoted in Figures 1a-c with vertical red arrows and in Table 2 with highlighted numbers.
The amplification boundaries covered the individual HERV-K14C sequences and their flanking regions. When a HERV-K14C locus was not observed in the orthologous region of primate genome, we can find a pre-integration site or intact structure of HERV-K14C that enabled us to estimate generation timing of Y chromosome. For each species tested, PCR products were not observed when DNA from female primates was used as template (data not shown).
Overall, the integration of HERV-K14C loci occurred in Old World monkey and hominoids approximately 25 million years ago (Figure 5). Most of HERV-K14C copies (5, 9/12, 15/19, 20, 21, 22, 24/25/26/29, and 27/28) resulted from insertion events that occurred after divergence of the hominoids from Old World monkeys. Specifically, copies 5, 15/19, 21, and 27/28 were only observed in humans and the samples from the great apes excluding orangutan suggesting that these insertions were occurred 10 million years ago.

The integration lineage of HERV-K14C copies in Y chromosome. The numbers correspond to the vertical red arrows and highlighted numbers in Figure 1 and Table 2, respectively. Hominoids: HU (human), CH (chimpanzee), GO (gorilla), OR (orangutan), and GI (gibbons); OWM (Old World monkeys): JM (Japanese monkey), RM (rhesus monkey); NWM (New World monkeys): NM (night monkey) and CM (common marmoset); ddH2O was used as a negative control.
In all copies, we only observed that 1 and 24/25/26/29 were an intact structure and pre-integration sites, respectively. The solitary LTR locus 1 (Table 2 no. 1) was present in not only the hominoid lineage, but an enlarged form of it was also found in the Old World monkey samples. Based on our sequencing data, the large product amplified from the Old World monkey samples had both LTRs and internal HERV sequences, and the sequence identity between the 5′ and 3′ LTR was quite high at approximately 82% (data not shown). Many HERV sequences have been lost their functional domains by internal recombination between the 5′ and 3′ LTRs leading to solitary LTR formation. In addition, a recombination between the 5′ and 3′ LTRs continues to occur in individuals of human population that has caused recent genetic variation in humans.27 In case of LTR locus 1, an HERV-K14C had integrated into the primate genome and then underwent a subsequent deletion event owing to homologous recombination between 5′ and 3′ LTRs.
Conversely, copies 24/25/26/29 on the b amplicon were unexpectedly small at approximately 384 bp with a gap of 7 bp) in Japanese and rhesus monkey genomes. In contrast, hominoids were revealed exact target size (976 bp); therefore, the LTR sequences copies 24/25/26/29 in the Old World monkey lineage seemed to have lost 585 bp. We attempted to confirm the sequences of unexpected product in the Old World monkeys. The sequencing results showed that a high degree of identity (86%) was found between flanking region of 24/25/26/29 and the small product. This observation suggested that 384 bp of small product was from a pre-integration site that existed before the integration of 24/25/26/29 LTRs. This data implied that the ancestral b amplicon has existed since the Old World monkey lineage. At least, the duplicated pairs that span P1 to P3 of the b amplicons were amplified after the divergence of hominoids and Old World monkey. Because the b amplicons reside in palindromic regions, it was quite difficult to amplify specific a locus from these regions. Thus, we could not clarify which of the b amplicons predated the insertion event and existed in the ancestral Y chromosome. Nevertheless, this investigation allowed us to analyze the complex evolutionary past of the Y chromosome.
The pair 15/19 was dispersed in P4, and this region was present only in human and gorilla consistent with an amplification of the inner boundaries of P4 from Page’s study.2 In addition, an identical pair of 9/12 on P5 was observed in the great apes, and the 27/28 locus on P1 identified an insertion site in gorilla, chimpanzee and human.
Obviously, orthologs of the HERV-K14C loci found in humans were detected in our closest evolutionary relatives, but we could not determine whether the integration time was consistent with the complete formation of the palindromic structure. In spite of that, our results demonstrated that at least an arm of P4 and of P5 already existed in the great ape lineage approximately 8–15 million years ago. In our result, most of loci were incapable of revealing a pre-integration site except locus 1 and loci 24, 25, 26, and 29. The majority of orthologs was not detected in samples from lesser apes, indicating that the genomic structure of human Y chromosome arose recently.
HERV-K14C was associated not only with palindromic structures but also with Y chromosomal transcripts. Using a computational approach, we screened human EST and mRNA databases with HERV-K14C consensus sequences to identify hybrid transcripts. As denoted in Figure 1d and Table 2, HERV-K14C provided gene regulatory regions to three transcripts. The Y chromosomal transcripts—AW966153, AK026367, and TTY13—were dispersed throughout the P4 and inverted repeat 2 (IR2) (Figure 1d).
Two identical copies of AW966153 were due to the duplication of P4 and solitary HERV-K14C LTRs from loci 15 and 19 provided the polyadenylation signals for these transcripts. Based on the integration lineage of LTRs on 15/19 locus, AW966153 was only expressed in the human and gorilla genomes. The 5′ LTR of locus 21 provided the promoter region for AK026367, and the first exon of this transcript was totally donated by HERV-K14C-related sequences. Using TRANSFAC® Professional version 10.1 with a strict threshold (core match: 1 and matrix match: 0.95∼1), we found five binding sites for multiple transcription factors (including GATA box, TFE, TGIF, and others) distributed on 5′ LTR of locus 21 which provided the transcription starting sites for AK026367.28 In addition, HERV-K14C sequences entirely provided the last two exons on TTY13 locus.
We used RT-PCR to examine the expression profiles of HERV-K14C-related transcripts in various human tissues (Figure 6). All of these HERV-K14C-related transcripts were exclusively expressed in human testis tissue. Based on genomic PCR result of loci 15/19, 21, and 22, these transcripts arose in the genome after divergence of the orangutan and gibbon lineages.

Expression profiles of HERV-K14C-related transcripts. GAPDH was used as internal marker, and ddH2O was used as a negative control.
Many researchers who study male infertility have investigated deletions of ampliconic sequences.29, 30, 31 For example, some studies reporting deletion between the distal arm of P1 to the proximal arm of P4 explained only those deletions that resulted from micro-deletions between direct repeat sequences that cause spermatogenic failure due to removal of Y chromosomal specific transcripts.32 In our investigation, three of HERV-K14C related transcripts spanned on between P4 and P1. The removal of HERV-K14C related transcripts has a potential possibility to be related with spermatogenic failure due to the deletion between P1 and P4 region. In this investigation, we could not fully elucidate the molecular role of HERV-K14C-related transcripts, but they were only detected in testis. In other words, HERV-related transcripts were not expressed in human testis when HERV-K14C was not present. Namely, the acquisition of testis-specificity could be attributed to the integration of HERV-K14C elements meaning that HERV-K14C gave a rise to transcriptome diversity in the Y chromosome during primate speciation.
In summary, we have estimated the evolutionary timing of the emergence of palindromic region of the Y chromosome. The orthologs of sequences flanking the HERV-K14C inserts were detected mostly after divergence of hominid lineage around 18–20 million years ago (Figure 7). Moreover, ancestral region of b amplicon existed in the Old World monkey lineage. We could not estimate the emergence date of both arms of each palindrome but one arm of P1.1/2, P3 and P4 was existed since gorilla lineage. Subsequently, they underwent the duplication event at least 10 million years ago. In our study, the integration timing of HERV-K14C presented orthologues of Y chromosome in primate genome that enabled to conjecture emergence timing of Y chromosomal sequences during primate evolution. Not only HERV-related sequences but also other transposable elements (TEs) should be useful tool to estimate evolutionary past of modern human Y chromosome as molecular clock. Moreover, these TEs have a possibility of different integration lineage that potentially have an effect on genomic differences between human and our closet relatives, implying that further studies of TEs lead us to understand a puzzled history of Y chromosome evolution.

Here, we suggested that the ancestral palindromic structure was constructed gradually as a pair of palindromes emerged during the evolutionary past and subsequently built up to its current structure of multiple palindromes due to duplication event that occurred in the great ape lineages. Analysis of Y chromosome sequences from the chimpanzee suggests that repeat sequences expanded and accumulated on the Y chromosome. In addition, this report suggested that the variation in sequence identity between repeat elements in human and chimpanzee was higher on the Y chromosome than it is on average across the genome implying that these repeated sequences changed more quickly.33
In our study, we investigated the orthologs of HERV-K14C integration sites that reflected the evolutionary past of the human Y chromosome. Because most of HERV family members emerged in the primate genome after the divergence of Old World and New World monkey lineages and some HERV-K family members still exhibit variability in human populations34, 35 HERVs may function as molecular clocks in the investigation of human evolution.
We aimed to verify the association between HERV-K14C and Y chromosome evolution. HERV-K14C-related sequences have been amplified during the evolution of the Y chromosome and contributed to genomic diversification of Y chromosome during great ape lineages.
References
Skaletsky, H., Kuroda-Kawaguchi, T., Minx, P. J., Cordum, H. S., Hillier, L., Brown, L. G. et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 423, 825–837 (2003).
Rozen, S., Skaletsky, H., Marszalek, J. D., Minx, P. J., Cordum, H. S., Waterston, R. H. et al. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature. 423, 873–876 (2003).
Kjellman, C., Sjogren, H. O. & Widegren, B. The Y chromosome: a graveyard for endogenous retroviruses. Gene. 161, 163–170 (1995).
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J. et al. Initial sequencing and analysis of the human genome. Nature. 409, 860–921 (2001).
Jurka, J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 16, 418–420 (2000).
Belshaw, R., Pereira, V., Katzourakis, A., Talbot, G., Paces, J., Burt, A. et al. Long-term reinfection of the human genome by endogenous retroviruses. Proc. Natl. Acad. Sci. USA 101, 4894–4899 (2004).
Lower, R., Lower, J. & Kurth, R. The viruses in all of us: characteristics and biological significance of human endogenous retrovirus sequences. Proc. Natl. Acad. Sci. USA 93, 5177–5184 (1996).
Sverdlov, E. D. Retroviruses and primate evolution. Bioessays. 22, 161–171 (2000).
Johnson, W. E. & Coffin, J. M. Constructing primate phylogenies from ancient retrovirus sequences. Proc. Natl. Acad. Sci. USA 96, 10254–10260 (1999).
Benit, L., Dessen, P. & Heidmann, T. Identification, phylogeny, and evolution of retroviral elements based on their envelope genes. J. Virol. 75, 11709–11719 (2001).
Mi, S., Lee, X., Li, X., Veldman, G. M., Finnerty, H., Racie, L. et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403, 785–789 (2000).
Landry, J. R. & Mager, D. L. Functional analysis of the endogenous retroviral promoter of the human endothelin B receptor gene. J. Virol. 77, 7459–7466 (2003).
Sin, H. S., Huh, J. W., Kim, D. S., Kang, D. W., Min, D. S., Kim, T. H. et al. Transcriptional control of the HERV-H LTR element of the GSDML gene in human tissues and cancer cells. Arch. Virol. 151, 1985–1994 (2006).
Sin, H. S., Huh, J. W., Kim, D. S., Kim, T. H., Ha, H. S., Kim, W. Y. et al. Endogenous retrovirus-related sequences provide an alternative transcript of MCJ genes in human tissues and cancer cells. Genes Genet. Syst. 81, 333–339 (2006).
Sverdlov, E. D. Perpetually mobile footprints of ancient infections in human genome. FEBS Lett. 428, 1–6 (1998).
Hughes, J. F. & Coffin, J. M. Evidence for genomic rearrangements mediated by human endogenous retroviruses during primate evolution. Nat. Genet. 29, 487–489 (2001).
Hughes, J. F. & Coffin, J. M. Human endogenous retroviral elements as indicators of ectopic recombination events in the primate genome. Genetics 171, 1183–1194 (2005).
Hughes, J. F. & Coffin, J. M. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: implications for human and viral evolution. Proc. Natl. Acad. Sci. USA 101, 1668–1672 (2004).
Cordaux, R. & Batzer, M. A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 10, 691–703 (2009).
Sun, C., Skaletsky, H., Rozen, S., Gromoll, J., Nieschlag, E., Oates, R. et al. Deletion of azoospermia factor a (AZFa) region of human Y chromosome caused by recombination between HERV15 proviruses. Hum. Mol. Genet. 9, 2291–2296 (2000).
Flockerzi, A., Burkhardt, S., Schempp, W., Meese, E. & Mayer, J. Human endogenous retrovirus HERV-K14 families: status, variants, evolution, and mobilization of other cellular sequences. J. Virol. 79, 2941–2949 (2005).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics. 21 (Suppl 1), i351–358 (2005).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic. Acids. Res. 25, 3389–3402 (1997).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic. Acids Res. 22, 4673–4680 (1994).
Tamura, K., Dudley, J., Nei, M. & Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599 (2007).
Mager, D. L. & Goodchild, N. L. Homologous recombination between the LTRs of a human retrovirus-like element causes a 5-kb deletion in two siblings. Am. J. Hum. Genet. 45, 848–854 (1989).
Matys, V., Fricke, E., Geffers, R., Gossling, E., Haubrock, M., Hehl, R. et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic. Acids Res. 31, 374–378 (2003).
Page, D. C., Silber, S. & Brown, L. G. Men with infertility caused by AZFc deletion can produce sons by intracytoplasmic sperm injection, but are likely to transmit the deletion and infertility. Hum. Reprod. 14, 1722–1726 (1999).
Choi, J., Koh, E., Matsui, F., Sugimoto, K., Suzuki, H., Maeda, Y. et al. Study of azoospermia factor-a deletion caused by homologous recombination between the human endogenous retroviral elements and population-specific alleles in Japanese infertile males. Fertil. Steril. 89, 1177–1182 (2008).
Kuroda-Kawaguchi, T., Skaletsky, H., Brown, L. G., Minx, P. J., Cordum, H. S., Waterston, R. H. et al. The AZFc region of the Y chromosome features massive palindromes and uniform recurrent deletions in infertile men. Nat. Genet. 29, 279–286 (2001).
Repping, S., Skaletsky, H., Lange, J., Silber, S., Van Der Veen, F., Oates, R. D. et al. Recombination between palindromes P5 and P1 on the human Y chromosome causes massive deletions and spermatogenic failure. Am. J. Hum. Genet. 71, 906–922 (2002).
Kuroki, Y., Toyoda, A., Noguchi, H., Taylor, T. D., Itoh, T., Kim, D. S. et al. Comparative analysis of chimpanzee and human Y chromosomes unveils complex evolutionary pathway. Nat. Genet. 38, 158–167 (2006).
Moyes, D., Griffiths, D. J. & Venables, P. J. Insertional polymorphisms: a new lease of life for endogenous retroviruses in human disease. Trends Genet. 23, 326–333 (2007).
Lebedev, Y. B., Belonovitch, O. S., Zybrova, N. V., Khil, P. P., Kurdyukov, S. G., Vinogradova, T. V. et al. Differences in HERV-K LTR insertions in orthologous loci of humans and great apes. Gene 247, 265–277 (2000).
Kim, H. S., Takenaka, O. & Crow, T. J. Isolation and phylogeny of endogenous retrovirus sequences belonging to the HERV-W family in primates. J. Gen. Virol. 80 (Pt 10), 2613–2619 (1999).
Acknowledgements
Supported in part by a Grant-in-Aid for Scientific Research (B) from the Japanese Ministry of Education, Science, Sports, and Culture (no.19390412). The authors also thank Ms E Saitoh and Ms N Kanno for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Sin, HS., Koh, E., Kim, DS. et al. Human endogenous retrovirus K14C drove genomic diversification of the Y chromosome during primate evolution. J Hum Genet 55, 717–725 (2010). https://doi.org/10.1038/jhg.2010.94
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.94
