
Abstract
Excessive cell proliferation and genetic changes such as loss of an allele (loss of heterozygosity (LOH)) or amplifications or deletions of parts of chromosomes (copy number variations (CNV)) are common findings in cancers. It is unknown whether these changes are also present in patients with overgrowth syndromes, although the presence of small-scale CNVs (such as duplication of 11p15 in Beckwith–Wiedemann syndrome), excessive cell proliferation and an increased frequency of tumors have all been reported in these patients. We present results of a genome-wide scan for LOH and CNV in Proteus syndrome (PS), a severely disfiguring overgrowth syndrome. We investigated CNV and LOH in DNA derived from affected and normal tissue samples from six PS patients using Affymetrix GeneChip Mapping 250 K Nsp high-density single-nucleotide polymorphism microarrays. Analysis revealed that LOH and CNVs were not common in PS. We attempted to validate selected CNVs detected by microarray analysis using quantitative genomic PCR, but the observed changes were not confirmed. These results suggest that large-scale genome-wide CNVs and LOH as seen in cancer syndromes are not characteristic findings in PS, although we cannot rule out the possibility that newer arrays with a higher number of probes could uncover smaller CNVs not detected in this study.
Similar content being viewed by others


Introduction
Proteus syndrome (PS) is a congenital and severely disabling condition characterized by asymmetrical tissue overgrowth that may affect any part of the body.1, 2 Relentless progression, vascular anomalies and epidermal naevi are some of the characteristic features of the syndrome.3, 4 Several types of tumors including haemangiomas, lymphangiomas and lipomas have also been reported in patients with PS.5, 6, 7
The causes of PS are unknown, but it has been hypothesized that the syndrome is a result of somatic mutations, which are lethal in the non-mosaic state.8 A genome-wide scan could reveal genetic lesions that might contribute to the development of PS. Specifically, chromosomal copy number variations (CNVs) and loss of heterozygosity (LOH) would be of particular interest. In cancer, LOH is frequently observed as a reduction to homozygosity of a lesion in the tumor DNA compared with the normal genomic DNA, often involving tumor suppressor genes. CNVs, which are also involved in cancer, are duplications or deletions in some parts of chromosomes ranging from one kb to several megabases in size.9
In cancer, investigation of LOH and CNV is usually performed by comparing DNA from lymphocytes or unaffected tissue to tumor DNA from the same person. Here, we report a genome-wide scan for CNV and LOH in PS. We used DNA derived from cultured fibroblasts taken from an affected part of the body of a child with PS paired with DNA derived from an unaffected region.
Materials and methods
Patients and DNA preparation
All patients were diagnosed with PS based on published diagnostic criteria10 (Table 1). Detailed clinical features in all of the patients, with the exception of patients 5 and 6, have been published.11 Vascular malformations were predominant in patient 5, whereas connective tissue nevus, thickening of the skin and subcutaneous tissue and facial phenotype were predominant in patient 6.
This study was approved by the Research Ethics Committee of each institution involved. DNA was extracted from blood samples or primary fibroblast cell cultures established from skin biopsies using commercially available DNA isolation kits. In addition, we used DNA extracted from a pair of normal/skin cancer cell lines obtained from American Type Culture Collection (ATCC, Manassas, VA, USA; catalog numbers CRL-7761 and CRL-7762) to investigate whether patterns of CNVs in PS and skin cancer were similar. The cell lines were established using samples taken from the same individual.
SNP microarrays
The genome-wide scan was performed using GeneChip Mapping 250 K Nsp single-nucleotide polymorphism (SNP) microarrays from Affymetrix (Santa Clara, CA, USA). A total of 262,264 SNPs covering the whole genome at approximately 3 kb intervals were genotyped. We also repeated microarray analysis on a pair of lung cancer cell line DNA (ATCC catalog numbers CCL-256D and CCL-256.1D) previously analyzed by Affymetrix. Before chip analysis, all samples were migrated on agarose gels to ensure lack of DNA degradation and their AD260/280 ratios were measured to ensure the absence of significant contamination with proteins; only high quality DNA samples were used for analysis.
Analysis of microarray data
The CNAT4.0.1 software, obtained from Affymetrix, was used to examine LOH and CNV status of PS patients in paired analysis by comparing the affected tissue to its normal counterpart or to the blood sample from the same person. We first used the following parameters (tumor parameters) recommended by the software manufacturer to detect CNV in tumor/normal paired samples: median normalization, genomic smoothing of 0.1 Mb, transition decay (TD) of 1 Mb, priors of 0.2 for Hidden Markow Model (HMM) and s.d. of 0.07 for CN state of 2 and 0.09 for the rest of CN states. In addition, outliers within the range of 1 kb were automatically removed. We then used the following modified parameters (micro deletion parameters) to detect micro deletions: median normalization, genomic smoothing of 15 kb, TD of 0.1 Mb, priors of 0.2 for HMM, s.d. of 0.13 for CN state of 2 and 0.15 for the rest of the CN states.
Quantitative genomic PCR
We used absolute quantitation using SybrGreenER (Invitrogen, Carlsbad, CA, USA) to confirm copy numbers obtained in microarray analysis following the manufacturer's instructions (see Supplementary Tables 1 and 2 for assay results and primer sequences).12, 13 This assay correctly identified the ratio of ALB to androgen receptor gene copy number (1.0 in six healthy females and 1.9 in six healthy males).
Results
We analyzed nine normal/affected sample pairs representing six patients (Table 1). Patient 4 had DNA available from two independent affected tissue samples whereas patients 2 and 3 had DNA isolated from two normal samples (tissue or blood). SNP call rates were over 99.3% in all samples (Table 1). To ensure reproducibility of the SNP microarrays, we re-analyzed one of the PS samples on a different date and obtained over 99% identical genotype calls. In addition, we genotyped a pair of lung tumor and blood sample from a patient previously analyzed by Affymetrix; 99.6% of our genotype calls were identical to those obtained by Affymetrix, indicating that these microarray experiments are essentially perfectly repeatable.
As we were interested in variants likely to have a significant role in PS development, as opposed to variations arising secondarily or as a result of culturing of cells, we only considered CNVs that were present in at least two patients; in addition, for patients who had more than one normal or affected tissue, the CNV had to be present in both comparisons for that patient to be considered positive. Finally, the type of CNV (that is, amplification vs. deletion) also had to be consistent between the two patients for the CNV to be considered biologically relevant to PS. Using the tumor detection parameters we found no LOH but detected 30 SNPs with CNV (that is, 23 SNPs with CNV seen in a single patient and seven SNPs with CNV shared by two patients; Supplementary Table 3). However, none of the seven SNPs shared by two patients fulfilled the above criteria.
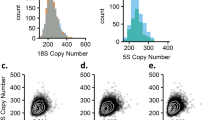
A total of 846 SNPs with CNV were detected using the microdeletion detection parameters (Figure 1 and Supplementary Table 4). Twenty four of these were seen in at least two patients, but 21 out of the 24 SNPs were observed in only one of the two normal/affected pairs in a patient, in which two pairs were genotyped. Two SNPs found in two patients showed amplification in one patient and deletion in the other, making them less likely to be variations with biological relevance in the development of the disease.

Paired whole-genome CNV analysis in Proteus syndrome and pairs of skin and lung cancer samples using CNAT. Numbers on the bottom row indicate chromosomes. Patient numbers are the same as in Table 1. We had more than one affected or normal sample from patients 2, 3 and 4, making it possible to perform two copy number analyses in these patients. Bars indicate where the CNV state of the affected tissue minus the CNV state of the normal tissue is non-zero (CVN state affected–CVN state normal=not zero). The amplifications or deletions shown are because of the observed differences in copy number between the DNA from affected and normal tissue, but the presence of an amplification or a deletion does not indicate whether the deviation from normal copy number is present in the normal or the affected tissue. The level of CNV in Proteus syndrome samples clearly does not resemble levels observed in tumors.
One interesting SNP overlapped by the SNX1 gene showed consistently reduced copy number in two patients including both normal/affected pairs from one patient by microarray analysis. However, quantitative genomic PCR of this SNP in our patients also showed normal copy number in all samples tested and did not confirm the CNV observed on the chip (Supplementary Table 1). In addition, 822 SNPs were observed to show CNV in only one patient; however, only 24 of 726 SNPs were seen in two sample pairs from a patient (3.3%), suggesting that a large proportion of the SNPs seen only in a single patient may represent background noise.
To verify the suitability of our analysis parameters to successfully detect LOH and CNV in our PS samples, we downloaded from the Affymetrix website microarray data from nine pairs of breast or lung tumor and their matched normal tissue DNA genotyped using same type of 250 K Nsp microarrays. We analyzed the data using the same set of conditions we used in CNAT to analyze the PS data and found widespread CNV and LOH in these as well as in our own skin tumor samples, despite finding limited CNV in PS (Figure 1).
Discussion
To date, little is known about the molecular basis of PS, but the most plausible underlying genetic cause is one or several somatic mutations in a subset of cells in genes involved in cell growth or proliferation, resulting in a mosaic state and localized phenotypes.8
Standard cytogenetic techniques did not detect any anomalies in the PS patients in our study (data not shown), and results from karyotype analysis by traditional methods and/or by comparative genomic hybridization of other cases of PS do not support the notion that large chromosomal defects are an important cause of PS (WD Foulkes and RS Houlston, unpublished data).
In this study, based on the rationale that physiological similarities between the uncontrolled cell proliferation observed in both tumors and PS may reflect underlying molecular similarities, we attempted to detect copy number changes in the genome of patients with PS in the hope that this approach might help to uncover underlying genetic mechanisms in the development and progression of the disorder. However, we observed no LOH and very few CNVs in PS. Although we cannot rule out the possibility that a higher density array might uncover changes that were missed in this study, we conclude that these types of genetic changes are not characteristic of disease progression in PS as they are in tumors and suggest that future efforts at elucidating the molecular mechanisms leading to the development of PS should be re-directed toward identifying possible epigenetic causes and performing next-generation sequencing of affected parts of the body in children with this relentlessly disfiguring disorder.
References
Furquim, I., Honjo, R., Bae, R., Andrade, W., Santos, M., Tannuri, U. et al. Proteus syndrome: report of a case with recurrent abdominal lipomatosis. J. Pediatr. Surg. 44, E1–E3 (2009).
Jamis-Dow, C. A., Turner, J., Biesecker, L. G. & Choyke, P. L. Radiologic manifestations of Proteus syndrome. Radiographics 24, 1051–1068 (2004).
Turner, J. T., Cohen Jr. M. M. & Biesecker, L. G. Reassessment of the Proteus syndrome literature: application of diagnostic criteria to published cases. Am. J. Med. Genet. A. 130A, 111–122 (2004).
Wiedemann, H. R., Burgio, G. R., Aldenhoff, P., Kunze, J., Kaufmann, H. J. & Schirg, E. The Proteus syndrome: partial gigantism of the hands and/or feet, nevi, hemihypertrophy, subcutaneous tumors, macrocephaly or other skull anomalies and possible accelerated growth and visceral affections. Eur. J. Pediatr. 140, 5–12 (1983).
Farajzadeh, S., Zahedi, M. J. & Moghaddam, S. D. A new gastrointestinal finding in Proteus syndrome: report of a case of multiple colonic hemangiomas. Int. J. Dermatol. 45, 135–138 (2006).
Debi, B., Nayak, S., Da, R. P. & Acharjya, B. Proteus syndrome. Indian J. Dermatol. Venereol. Leprol. 71, 357–359 (2005).
Hotamisligil, G. S. Proteus syndrome and neurofibromatosis. Neurofibromatosis 2, 339–340 (1989).
Happle, R. Cutaneous manifestation of lethal genes. Hum. Genet. 72, 280 (1986).
Cook, E. H. & Scherer, S. W. Copy-number variations associated with neuropsychiatric conditions. Nature 455, 919–923 (2008).
Biesecker, L. G., Happle, R., Mulliken, J. B., Weksberg, R., Graham, J. M. Jr., Viljoen, D. L. et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am. J. Med. Genet. 84, 389–395 (1999).
Thiffault, I., Schwartz, C. E., Der Kaloustian, V. & Foulkes, W. D. Mutation analysis of the tumor suppressor PTEN and the glypican 3 (GPC3) gene in patients diagnosed with Proteus syndrome. Am. J. Med. Genet. A 130, 123–127 (2004).
Barrois, M., Bièche, I., Mazoyer, S., Champème, M. H., Bressac-de Paillerets, B. & Lidereau, R. Real-time PCR-based gene dosage assay for detecting BRCA1 rearrangements in breast-ovarian cancer families. Clin. Genet. 65, 131–136 (2004).
Kindich, R., Florl, A. R., Jung, V., Engers, R., Müller, M., Schulz, W. A. et al. Application of a modified real-time PCR technique for relative gene copy number quantification to the determination of the relationship between NKX3.1 loss and MYC gain in prostate cancer. Clin. Chem. 51, 649–652 (2005).
Acknowledgements
This research was supported by grants from the South Carolina Department of Disabilities and Special Needs (SCDDSN) (to CES), Cancer Research Society Inc. (to WDF) and a Montreal Centre for Experimental Therapeutics in Cancer/CIHR fellowship (to AY). We thank La Fond de la Recherche en Santé du Québec (FRSQ) for institutional grants supporting our facilities.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Yilmaz, A., Hamel, N., Schwartz, C. et al. A genome-wide analysis of loss of heterozygosity and chromosomal copy number variation in Proteus syndrome using high-density SNP microarrays. J Hum Genet 55, 627–630 (2010). https://doi.org/10.1038/jhg.2010.70
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.70
Keywords
This article is cited by
-
Von der Diagnostik zur intraoperativen Navigation
Der MKG-Chirurg (2012)
