
Abstract
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is an autosomal, recessively inherited disease caused by mutations in the MLC1 gene. Most of the previously published studies have been carried out in ethnic populations other than the Chinese. In this study, the analysis of clinical features and MLC1 mutation screening were performed in 13 Chinese patients for the first time. A total of 10 MLC1 mutations were identified in these patients, including five novel missense mutations (c.65G>A, p.R22Q; c.95C>T, p.A32V; c.218G>A, p.G73E; c.823G>A, p.A275T; c.832T>C, p.Y278H), one novel splicing mutation (c.772-1G>C in IVS9-1), one novel small deletion (c.907_930del, p.V303_L310del), one known nonsense mutation (c.593delCTCA, p.Y198X) and two known missense mutations (c.206C>T, p.S69L; c.353C>T, p.T118M). Mutation c.772-1G>C in IVS9-1, accounting for 27.3% (3/11) of the total number of genetically confirmed patients found in this study, is thus a putative hot-spot mutation in the present study group. The existence of a unique MLC1 mutation spectrum in Chinese MLC patients was shown. A systemic study to assess the mutation spectra in different populations should be undertaken.
Similar content being viewed by others

Introduction
Megalencephalic leukoencephalopathy with subcortical cysts (MLC, OMIM 604004) is a rare vacuolating myelinopathy characterized by early-onset macrocephaly presenting at birth or during the first year of life, often accompanied by the occurrence of motor dysfunction provoking severe walking disability, onset of seizure and usually late onset of mild mental deterioration.1 Severity ranges from independent walking for only a few years to independent walking into the fifth decade. Some patients die in their teens or 20s, whereas others stay alive into their 40s. The features seen in cranial magnetic resonance imaging (MRI) are unique and very helpful in the clinical diagnosis of MLC.1, 2 MRI typically shows diffuse white-matter abnormalities with subcortical cysts in the tips of the temporal lobes and in frontoparietal subcortical areas.
MLC, inherited in an autosomally recessive manner, is a heterogeneous genetic condition resulting from gene defects in MLC1, first identified in 2001.3 The MLC1 (GenBank NM_015166) maps to chromosome band 22q13.3; it covers ∼20 kb of genomic DNA and includes 12 exons.3 To date, about 68 MLC-relevant mutations of MLC1 have been identified in patients of various ethnic backgrounds (human gene mutation database). However, families unlinked to the MLC1 locus have been reported,3 and the existence of at least one other locus has been hypothesized.4, 5 Chinese patients with genetic confirmative diagnosis of MLC have never been reported before. In this study, we analyzed 13 Chinese patients and identified 10 mutations, including seven novel mutations in MLC1.
Materials and methods
Diagnostic criteria of MLC
Clinical diagnosis was mainly based on the criteria proposed by van der Knaap et al. (http://www.genetests.org), including the following: (1) Macrocephaly is present at birth or, more commonly, develops within the first year of life in all individuals. (2) Early development is normal or mildly delayed. (3) The clinical presentation is slow deterioration of motor functions, with cerebellar ataxia and mild spasticity usually starting in early childhood or later. Mental decline occurs later and is much milder than motor decline. Most individuals have epileptic seizures that are usually easily controlled with medication. (4) Neurological signs mainly consist of cerebellar ataxia and subsequent pyramidal dysfunction. (5) Criteria for being eligible for cranial MRI include the following: (i) The cerebral hemispheric white matter is diffuse, abnormal and mildly swollen, and the white-matter swelling decreases and cerebral atrophy ensues over time. (ii) Subcortical cysts are almost invariably present in the anterior-temporal region and often in the frontoparietal area; the subcortical cysts may increase in size and number. (iii) Central white-matter structure including the corpus callosum, internal capsule and brain stem are better preserved. Overall 13 patients who met the criteria were included.
Clinical investigation
All patients underwent extensive clinical investigation, including: (1) physical examination; (2) blood tests for serum levels of electrolytes, ammonia, lactate and pyruvate, and the assessment of liver and renal functions; (3) assessment of the enzymatic activities of arylsulfatase A and galactocerebroside β-galactosidase in peripheral white blood cells; (4) urinary screening for disorders of amino acid, organic acid and fatty acid metabolism; and (5) cranial MRI.
DNA preparation and mutation screening
Informed consent was obtained from all families. Genomic DNA was extracted from peripheral leukocytes. Mutation screening for MLC1 was carried out in each patient. In total, 12 exons of MLC1, including flanking introns, were amplified. The sequences of the primers and the conditions for PCR are available on request. DNA sequencing was carried out using the ABI PRISM BigDye Terminator cycle Ready Reaction sequencing kit (Applied Biosystems, Carlsbad, CA, USA) on an applied Biosystems 3100 automatic DNA sequencer. For novel mutations found in the patients, the corresponding amplicons were amplified from 170 control samples, followed by sequencing and restriction endonuclease digestions.
Results
Clinical features of Chinese MLC patients
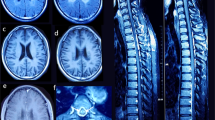
The study group consisted of unrelated patients, except for two pairs of siblings (patients 6 and 7 and patients 10 and 11), with eight boys and five girls from 11 core families. Their ages ranged from 1 year and 10 months to 13 years. The initial presentation of all individuals was macrocephaly, which began to appear between birth and 1 year of age. Independent sitting and walking were achieved in all but three cases, delayed from mildly to moderately. The exceptions were patients 4 and 8, who could not walk even at 2 years and 9 months and 4 years of age, respectively, and patient 13, who was only 1-year old. Their cognitive functions were relatively better preserved except for patients 3 and 5, with mild and moderate mental retardation, respectively. Among the 13 patients, two patients had occasional seizures during the course, patient 4 had only one febrile seizure, patient 5 had four afebrile seizures and one patient had recurrent seizures. Physical examination mainly showed large head circumference, low muscle tone and symmetric pyramidal tract signs. All individuals were children of non-consanguineous parents. Family history was negative in all but the sibling pairs: patients 6 and 7 and patients 10 and 11. The cranial MRI showed diffuse long T1 and T2 abnormal signal changes in cerebral white matter, with water-like subcortical cysts in anterior-temporal regions in all cases (Figure 1). All other clinical infestations were unrevealing. The major clinical features of these patients are summarized in Table 1.

Cranial magnetic resonance imaging (MRI) findings in patient 1. (a) Shows diffuse and abnormal swollen white-matter signals in T2-weighted images. (b, c) Demonstrate subcortical cysts in the anterior-temporal area (arrows) in T1-weighted images.
MLC1 mutation screening analysis
MLC1 mutations were identified in 11 of the 13 patients (84.6%), consisting of five novel missense mutations (c.65G>A, p.R22Q; c.95C>T, p.A32V; c.218G>A, p.G73E; c.823G>A, p.A275T; c.832T>C, p.Y278H), one novel splicing mutation (c.772-1G>C in IVS9-1), one novel small deletion (c.907_930del, p.V303_L310del), one known nonsense mutation (c.593delCTCA, p.Y198X) and two known missense mutations (c.206C>T, p.S69L; c.353C>T, p.T118M). The seven novel mutations were not detected in 170 normal control samples. Mutation c.772-1G>C in IVS9-1 accounted for 27.2% (3/11) of the total number of genetically confirmed patients and 21.1% (4/19) of total number of mutations found in this study. Patients were compound heterozygous or homozygous, with the mutations inherited from their parents. In three patients, mutations were identified only in one allele and a reported single-nucleotide polymorphism c.*48A>T in the 3′-untranslated region (patients 4 and 9) and a reported single-nucleotide polymorphism c.512G>T (p.C171P) (patient 5) in another allele. For sibling patients 6 and 7, two heterozygous mutations were revealed. One homozygous mutation was found in sibling patients 10 and 11. Two reported single-nucleotide polymorphisms were found in patients 12 and 13. All the MLC1 mutations found in this study are shown in Table 2.
Discussion
MLC is an autosomal, recessive neurological disorder in children characterized clinically by early-onset macrocephaly, often in combination with mildly delayed gross motor development and seizures, gradual onset of ataxia, spasticity and some additional pyramidal findings, usually with a late onset of mild mental deterioration. Seizures usually occur from an early age but can be well controlled by treatment with antiepileptic drugs. MRI shows a swollen brain with diffusely abnormal cerebral white matter and subcortical cysts in the anterior-temporal and frontoparietal regions. All 13 Chinese patients who met the criteria proposed by van der Knaap et al.1, 6 had the typical clinical features.
All 13 Chinese patients in the present study had early-onset macroencephaly, with onset before 1 year of age. Most of them presented mildly to moderately delayed motor development. In all, 2 out of the 13 patients revealed mental retardation. Seizures occurred in patients 4, 5 and 12. Patients 4 and 5 did not receive anticonvulsant drug therapy, and seizures were well controlled in patient 12 by drug therapy. Diffuse abnormal cerebral white matter and subcortical cysts in the cranial MRI, the hallmark of MLC, were seen in all our patients. Thus, all 13 patients in this study met the criteria proposed by van der Knaap et al. according to their clinical features and MRI findings.
Mutations in MLC1 were identified in 11 of the 13 patients (84.6%) with MLC, whereas only two single-nucleotide polymorphisms were found in the two remaining MLC patients. This result is similar with the reported incidence of approximately 80% of mutations in MLC1 found by van der Knaap.7 Further studies in large cohorts are still needed to provide insight into the frequency of positive mutations.
To date, about 68 MLC-relevant mutations of MLC1 have been identified in patients from various ethnic backgrounds (human gene mutation database), but no findings from a Chinese population have been reported previously. In this study, 10 MLC1 mutations were found, including five novel missense mutations (c.65G>A, p.R22Q; c.95C>T, p.A32V; c.218G>A, p.G73E; c.823G>A, p.A275T; c.832T>C, p.Y278H), one novel splicing mutation (c.772-1G>C in IVS9-1), one novel small deletion (c.907_930del, p.V303_L310del), one known nonsense mutation (c.593delCTCA, p.Y198X) and two known missense mutations (c.206C>T, p.S69L; c.353C>T, p.T118M). Seven novel mutations were not detected in 170 normal control samples. All mutations occurred in transmembrane domains 1–7 of the eight domains in the MLC protein.8, 9 These caused changes in the chemical properties of the residues, that is, from basic (R) to amide (Q) for mutation p.R22Q and from hydrophobic (G and A) to acidic (E) and polar (T) for mutations p.G73E and p.A275T, respectively. This may result in an alteration of the protein's secondary structure, which may affect protein function. The splicing mutation c.772-1G>C in IVS9-1 changes the splice-acceptor AG sequence of intron 9 to AC. This mutation most likely leads to skipping of exon 9, which will result in a deletion of 19 amino acids from the protein.
One known heterozygous nonsense mutation, c.593delCTCA (p.Y198X) in exon 7, results in the translation of only 198 out of the total 377 amino acids. This may affect the function of the MLC1 protein. Mutation p.Y198X was first reported in two Croatian sibling patients by Leegwater et al.,8 who reported that the 4-bp deletion led directly to a stop codon at the exon 7/exon 8 junction and did not affect the efficiency of the exon 7/intron 7 splicing donor predicted by the program SPLICEVIEW. Another known missense mutation, p.S93L, has been reported fairly frequently in Japanese individuals.10
Altogether, the 10 novel and reported MLC1 mutations found in the present study seemed to be functionally important because they may affect amino acids conserved among rat, mouse and human proteins, and amino-acid positions of the 10 mutations were distributed over a wide range from the N-to the C-terminus of the MLC1 protein. Further study is needed to provide insight into the effects of these mutations on the function of MLC1.
In contrast with reports on MLC patients of other ethnic origins, c.135_136insC (p.C46LfsX34) was a common mutation found in the East-Indian population,10, 11, 12 p.G59E was relatively frequent among Libyan Jews and mutation S93L appears in Japanese individuals.10 In this study of Chinese patients, a spectrum of MLC1 mutations different from that in earlier reports was found. The above commonly reported mutations were not identified in any of our patients, except for p.S93L, which was found in one allele in one patient. However, the splicing mutation c.772-1G>C was shared by three of our patients, accounting for 27.2% (3/11) of the total number of genetically confirmed patients and 21.1% (4/19) of the total number of mutations found in this study. This may be a hot-spot mutation in Chinese MLC patients, but this speculation must be verified by detection in more Chinese MLC patients screened for mutations in MLC1.
Therefore, from our preliminary data, it may be inferred that Chinese MLC patients may have a unique spectrum of MLC1 mutations, different from that of other ethnic populations; this knowledge will be helpful to set up a mutation screening strategy for MLC in China.
The genetic confirmation of MLC in Chinese patients is reported here for the first time, with seven novel mutations in identified MLC1. A unique MLC1 mutation spectrum was found in Chinese patients. To assess the incidence of MLC and the mutation spectra in different populations, a systemic study must be carried out. Further functional studies of the MLC1 mutations found in this study are under way in our group to delineate the correlation between MLC1 gene defects and the pathogenesis of MLC.
Accession codes
References
van der Knaap, M. S., Barth, P. G., Stroink, H., van Nieuwenhuizen, O., Arts, W. F., Hoogenraad, F. et al. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann. Neurol. 37, 324–334 (1995).
Morita, H., Imamura, A., Matsuo, N., Tatebayashi, K., Omoya, K., Takahashi, Y. et al. MR imaging and 1H-MR spectroscopy of a case of van der Knaap disease. Brain. Dev. 28, 466–469 (2006).
Leegwater, P. A., Yuan, B. Q., van der Steen, J., Mulders, J., Konst, A. A., Boor, P. K. et al. Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am. J. Hum. Genet. 68, 831–838 (2001).
Blattner, R., von Moers, A., Leegwater, P. A., Hanefeld, F. A., van der Knaap, M. S. & Kohler, W. Clinical and genetic heterogeneity in megalencephalic leukoencephalopathy with subcortical cysts (MLC). Neuropedia. 34, 215–218 (2003).
Patrono, C., di Giacinto, G., Eymard-Pierre, E., Santorelli, F. M., Rodriguez, D., De Stefano, N. et al. Genetic heterogeneity of megalencephalic leukoencephalopathy and subcortical cysts. Neurol. 61, 534–537 (2003).
van der Knaap, M. S., Smit, L. M., Barth, P. G., Catsman-Berrevoets, C. E., Brouwer, O. F., Begeer, J. H. et al. Magnetic resonance imaging in classification of congenital muscular dystrophies with brain abnormalities. Ann Neurol. 42, 50–59 (1997).
van der Knaap, M. S., Lai, V., Kohler, W., Salih, M. A., Fonseca, M. J., Benke, T. A. et al. Megalencephalic leukoencephalopathy with cysts without MLC1 defect. Ann. Neurol. 67, 834–837 (2010).
Ilja Boor, P. K., de Groot, K., Waisfisz, Q., Kamphorst, W., Oudejans, C. B., Powers, J. M. et al. MLC1: a novel protein in distal astroglial processes. J. Neuropathol. Exp. Neurol. 64, 412–419 (2005).
Leegwater, P. A., Boor, P. K., Yuan, B. Q., van der Steen, J., Visser, A., Konst, A. A. et al. Identification of novel mutations in MLC1 responsible for megalencephalic leukoencephalopathy with subcortical cysts. Hum. Genet. 110, 279–283 (2002).
Tsujino, S., Kanazawa, N., Yoneyama, H., Shimono, M., Kawakami, A., Hatanaka, Y. et al. A common mutation and a novel mutation in Japanese. J. Hum. Genet. 48, 605–608 (2003).
Singhal, B. S., Gursahani, R. D., Udani, V. P. & Biniwale, A. A. Megalencephalic leukodystrophy in an Asian Indian ethnic group. Pediatr. Neurol. 14, 291–296 (1996).
Ben-Zeev, B., Gross, V., Kushnir, T., Shalev, R., Hoffman, C., Shinar, Y. et al. Vacuolating megalencephalic leukoencephalopathy in Israeli patients. J. Child. Neurol. 16, 93–99 (2001).
Acknowledgements
We thank the patients and their parents for their support and cooperation. This work was supported by the National Key Research Project ‘11-5’ (No. 2006BAI05A07), ‘973’ Project of the Science and Technology Ministry of China (No. 2007CB5119004), the National Natural Science Foundation of China (30973227), the International Collaborative Genetic Research Training Grant (NIH/FIC, No. D43 TW06176)) and the Peking University First Hospital Returned Overseas Doctor Research Foundation.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Wang, J., Shang, J., Wu, Y. et al. Identification of novel MLC1 mutations in Chinese patients with megalencephalic leukoencephalopathy with subcortical cysts (MLC). J Hum Genet 56, 138–142 (2011). https://doi.org/10.1038/jhg.2010.146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.146
Keywords
This article is cited by
-
Carrier burden of over 300 diseases in Han Chinese identified by expanded carrier testing of 300 couples using assisted reproductive technology
Journal of Assisted Reproduction and Genetics (2023)
-
A Commentary on Identification of novel MLC1 mutations in Chinese patients with megalencephalic leukoencephalopathy with subcortical cysts (MLC)
Journal of Human Genetics (2011)
