
Abstract
We examined the influence that rare variants and low-frequency polymorphisms in the cancer candidate gene CCND1 have on the development of multiple intestinal adenomas and the early onset of colorectal cancer. Individuals with <100 multiple polyps and patients with colorectal cancer diagnosed before 50 years of age were recruited in UK, and screened for sequence changes in the coding and regulatory regions of CCND1. A set of about 800 UK control individuals was genotyped for the variants discovered in the cases. Variants in the promoter, intron-exon boundaries and untranslated regions of the CCND1 gene had higher frequencies in cases than in controls. Five of these variants were typed in a set of French multiple adenoma and early-onset patients, who also showed higher allele frequencies than UK controls. When pooled together, variants with frequencies lower than 1% conferred an increased risk of disease that was significant in the multiple adenoma group (odds ratio (OR) 2.2; 95% confidence interval, 1.1–4.4; P=0.03). Most variants had a putative functional effect when assessed in silico. We conclude that rare variants of CCND1 are risk factors for colorectal cancer, with considerably larger effects than common polymorphisms, and as such should be systematically evaluated in susceptibility studies.
Similar content being viewed by others

Introduction
The CCND1 gene is located on chromosome 11q13 and encodes the protein cyclin D1 that regulates cell cycle progression from G1 to S phase during cell division through its interactions with the cyclin-dependent kinases.1 Increased cyclin D1 expression has been reported as an early event in colorectal tumorigenesis2 and has been observed in other cancer types, including prostate, breast, lung and endometrial carcinomas.3, 4 Prior work has shown, however, that CCND1 is not essential for the development of colorectal cancer, although it may act as a modifier of disease severity.5
Most association studies of CCND1 have so far focused on the common and functionally significant G870A (P241P, rs9344) polymorphism, which affects splicing by eliminating a donor site at the end of exon 4. However, results correlating this polymorphism with cancer risk have been inconsistent. A recent meta-analysis of 60 published case-control studies has shown that, overall, individuals with the GA or AA genotype exhibited a 1.1- to 1.2-fold increased risk of developing cancer compared with individuals with the GG genotype.6 With respect to colorectal cancer in particular, subjects with hereditary nonpolyposis colorectal cancer who carry the rs9344 polymorphism have been reported to acquire the disease at an earlier age.7, 8, 9 Also, carriers of the A allele appear to be more frequent among individuals who developed non-syndromic colorectal cancer before the age of 60,10 subjects with familial colorectal cancer11, 12 and affected women.13, 14 Other reports, however, do not subscribe to rs9344 genotype being a modifier of the colorectal cancer phenotype.15, 16
Given the low odds ratios (OR) associated with common variants, such as rs9344, we have argued that genetic risk factors underlying complex diseases are more likely to be due to functionally relevant rare variants with moderate penetrance that will considerably increase susceptibility and will, therefore, sometimes justify prophylactic interventions.17, 18, 19 We defined rare variants as those having higher frequencies than rare severe effect, clearly familial, mutations but lower frequencies than polymorphisms. Thus, rare variants will generally be in the frequency range between 0.1 and 1%. We consider low-frequency variants to be those with frequencies between 1 and 5%, which are not normally used in standard association studies. Following the strategy we previously proposed,19 we have screened the regulatory and coding regions of CCND1 in individuals with multiple adenomas and patients with early-onset colorectal cancer recruited in the UK clinics. A few of the variants found this way were examined in a group of similarly ascertained French patients. We sought to assess the impact that collections of variants with gene frequencies lower than 1%, and between 1 and 5% have on the onset and progression of colorectal cancer, and what role, if any, the rs9344 polymorphism has in the pathogenesis of this disease.
Subjects and methods
Subjects
The UK patient group consisted of 112 individuals with 3–100 histologically proven synchronous or metachronous adenomatous polyps,18 and 44 individuals with colorectal cancer diagnosed before 50 years of age. A total of 38 individuals with early-onset disease were obtained through the VICTOR clinical trial, a Phase III double-blind placebo controlled study of rofecoxib in Dukes stage B or C colorectal cancer patients following potentially curative therapy, whereas the remaining six were recruited through the John Radcliffe and Churchill hospitals’ gastrointestinal clinics. With the exception of one Black Caribbean and one Indian individual, ethnic origin was White British for all UK patients for whom information was available. Non-white individuals were excluded from further analysis. No patient fulfilled the criteria for familial adenomatous polyposis, autosomal recessive MYH-associated polyposis or hereditary nonpolyposis colorectal cancer on clinical grounds.18 Some of these patients had already been screened for germline mutations in the APC and MYH genes during previous studies.20, 21
In addition, we collected samples from 131 French patients, 75 with multiple adenomas and 56 with early-onset colorectal cancer, who were recruited in the Department of Digestive Surgery at the Hôpital Saint-Antoine in Paris. All patients who underwent a colectomy or total coloproctectomy for colorectal cancer or polyposis were selected for the study a priori. Those diagnosed with colorectal cancer before the age of 50 years or with more than three polyps detected after 2005, were referred for a consultation with the geneticist. Immunohistochemical staining to determine loss of expression of the genes MLH1 and MSH2 and microsatellite status was performed for all patients with early-onset colorectal cancer. Sequencing of the entire MYH and APC genes was carried out in patients with multiple adenomatous polyps. Only patients with no evidence of hereditary nonpolyposis colorectal cancer, autosomal recessive MYH-associated polyposis or familial adenomatous polyposis were included in the current study. No ethnic identification was available for the French patients.
All UK and French cases had histological confirmation of adenomatous polyps, but the precise number of polyps was not determined for all of them. For 24 UK and 14 French adenoma patients, only ‘multiple’ was recorded.
Within both the UK and French patient groups, individuals with attenuated familial adenomatous polyposis may be included, as they were not purposefully eliminated from the study.
Controls comprised of 866 individuals, collected in 10 different regions across the UK, as part of the People of the British Isles study (see website link below), and were unselected with respect to disease status.
Blood samples from cases and controls and clinicopathological information from patients were collected with individual informed consent and local ethical committee approvals.
DNA extraction and processing
Genomic DNA for patient samples was extracted from peripheral venous blood using standard techniques. The People of the British Isles study control blood samples were transported at room temperature to the laboratory, where the peripheral blood lymphocytes were separated under sterile conditions22 within 2 days of collection. DNA was prepared from the 10 ml blood residue, remaining after sterile separation using either magnetic beads (GeneCatcher, Invitrogen, Carlsbad, CA, USA) or spin columns (Qiagen, Valencia, CA, USA). DNA concentration was determined using PicoGreen23 and normalized for genotyping to 25 ng μl−1. Samples from UK cases underwent whole genome amplification because of limited volumes and amounts of genomic DNA. We used the Repli-g Mini kit (Qiagen) that implements a multiple displacement amplification reaction to generate up to 10 μg of DNA per 50 μl reaction from a starting amount of at least 10 ng of genomic template. Genomic DNA from French cases and UK controls was used for genotyping.
PCR amplification
All exons, 5′untranslated region (UTR), 3′UTR, intron-exon boundaries and about 1.5 kb of the CCND1 promoter were screened, covered by a total of 21 PCR fragments (Supplementary Table 1). DNA amplification was carried out in 50 μl reactions with a final concentration of 1X PCR Gold buffer, 200 μM dNTPs and 0.5 μM of each primer. AmpliTaq Gold (2 U) (Applied Biosystems, Foster City, CA, USA), 1.5–2.5 mM of MgCl2 and 20–50 ng of genomic or whole-genome amplified DNA were used per reaction. Cycling conditions basically consisted of an initial denaturing step at 95 °C for 10 min, followed by 35 cycles at 95 °C for 25 s; annealing temperature 55 °C–65 °C, 35 s; 72 °C, 30 s; and a final elongation step at 72 °C for 5 min. Agarose gels (2%) were run to verify the successful amplification of each fragment.
Mutation analysis
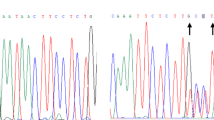
Mutation screening in UK patients was performed using a WAVE DNA fragment analysis dHPLC system with UV detection (Transgenomic, Omaha, NE, USA). Temperature gradients were designed using the WaveMaker software (Transgenomic) to obtain fragment-melting profiles. PCR products, of sizes between 234 and 522 bp, were denatured for 5 min at 94 °C, then gradually reannealed by decreasing the temperature to 25°C for 30 min to form homo and/or heteroduplexes. PCR products were subsequently eluted through an acetonitrile gradient at 0.9 ml min−1 over 6.8 min, at one or two different temperatures, selected according to their melting profiles. The column mobile phase consisted of buffer A, a 0.1 M triethylammonium acetate solution at pH 7, and buffer B, a 0.1 M triethylammonium acetate solution containing 25% acetonitrile at pH 7. The retention time of the eluate was registered by the ultraviolet detector at 260 nm. Under these conditions, invariant DNA fragments elute as a single peak, whereas variant fragments, which contain mixtures of homoduplex and heteroduplex DNAs, elute as two to four peaks or as a single peak with a shoulder.
DNA sequencing and genotyping
Cases with heteroduplex peaks, and several individuals with only homoduplex peaks were sequenced to identify the genetic variants. Each PCR product was purified following the EXO-SAP protocol and submitted to the Weatherall Institute of Molecular Medicine central facility for direct sequencing. Whenever possible, both strands were sequenced. Sequences were analyzed using Sequence Scanner version 1 (Applied Biosystems) and compared with the CCND1 GenBank entries NM_053056 and NT_167190.
All newly discovered variants were verified by genotyping the UK cases. Controls were then genotyped for variants identified and validated in patients. Two variants (CCND1–3 and 30) were genotyped in a subset of only 222 People of the British Isles study controls. Because the screening was not fully successful for exon 2 and the distal 5′-upstream fragments, we selected 12 additional CCND1 variants from the single nucleotide polymorphism database (dbSNP) (⩽3% minor allele frequency in HapMap European population, when reported) located in these regions, and typed them in both cases and controls. French patients were genotyped for a subset of five variants: two promoter variants (CCND1–3, 7), one 5′UTR variant (CCND1–19), one coding SNP (CCND1–21) and one 3′UTR SNP (CCND1–30). Genotyping of all variants, including rs9344, was done using the Sequenom MassArray technology, namely matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry and the iPLEX Gold assay (Sequenom Inc., San Diego, CA, USA).
Statistical analysis
Hardy–Weinberg equilibrium was tested for in controls using a χ2 goodness-of-fit test. Variant differences between cases and controls were assessed using Fisher's exact test. Two-sided P-values below 0.05 were considered statistically significant. Heterogeneity between UK multiple adenoma and early-onset cases was evaluated with a contingency table approach implemented in the program PowerMarker version 3.25 (Liu and Muse24). Odds ratios and 95% confidence interval were estimated with the software package SPSS version 16.0 (SPSS, Chicago, IL, USA).
Linkage disequilibrium patterns across CCND1 were examined using the program Haploview version 4.1,25 and by counts in 2 × 2 tables and their statistical assessment.
Functional in silico analysis of CCND1 variants to identify changes in transcription factor binding and alternative splicing sites were carried out using the web programs AliBaba 2.1 and Human Splicing Finder,26 respectively. The effect of 5′UTR and 3′UTR variants was analyzed using the online database UTRdb. Additionally, we used the GenEpi toolbox27 to examine conservation across species, as well as disruption of microRNA target sequences in the 3′UTR.
Results
The characteristics of both sets of patients and controls are summarized in Table 1 All CCND1 variants were in Hardy–Weinberg equilibrium in controls. A list of all variants, either first identified by dHPLC and/or selected from dbSNP, is given in Supplementary Table 2, and patient and control gene frequency data are given in Supplementary Table 3.
Stratifying the cases into multiple adenoma and early-onset groups (see Table 2) suggested that there might be some differences in the variant frequencies between the UK and French groups, notably for the CCND1–3 variant. Heterogeneity test results showed that there were no significant differences in allele frequencies per locus between the UK multiple adenoma and early-onset subgroups. This justifies combining the sets of UK patients for an assessment of overall association patterns.
Rare, low frequency and common variants
Overall 20 variants were typed in UK cases and controls, of which 10 were detected through mutation analysis and 10 were selected directly from dbSNP. Four additional variants were found by dHPLC screening, but could not be validated because of technical issues with the Sequenom assays (CCND1–23, 24, 25, 26). Similarly, two further dbSNP variants were examined in patients and were not subsequently analyzed in controls because they could not be successfully typed (CCND1–5, 6). There were three common variants detected, with gene frequencies equal to or higher than 10% in the HapMap European population (CEU), one of which was rs9344 (CCND1–21, 23, 30). The other two were not included in the analysis. Another common polymorphism located in exon 5, rs7177, was excluded as well. Of all the variants successfully genotyped in patients and controls, five sites were invariant in both and these were also excluded from analysis (CCND1–1, 2, 11, 14, 16). Three variants were monomorphic only in controls (CCND1–19, 22, 28), and one was invariant only in cases (CCND1–15), and these were included in the analysis, which therefore involved 14 variants in total, including rare, low frequency and common variants. In all, 6 out of the 14 variants uncovered by dHPLC screening of the UK samples were not present in dbSNP (CCND1–17, 18, 22, 24, 25, 26). Three variants (CCND1–13, 20, 29) had reported CEU gene frequencies between 3 and 5%, and two variants (CCND1–16, 27) had CEU gene frequencies lower than 1% (see Supplementary Table 2 for more details about the variants detected, genotyped and subsequently analyzed).
Association analysis
In an analysis with relatively small numbers, it is not expected that individual variants will show significant effects unless they are associated with large ORs. Nevertheless, one rare variant (CCND1–7) did give a significant P-value for the difference between UK cases (combining the early onset and multiple adenoma sets) and controls (OR 3.7; 95% confidence interval 1.2–11.8; P=0.03) (Table 3).
When we examined all rare variants together there was a significant increase in risk (OR⩾2) for the UK multiple adenoma group as well as for all UK cases (Table 4). There was also an increase in risk for the early-onset group considered on its own, but it did not reach significance presumably because of the small sample size.
There is strong linkage disequilibrium (LD) in the region between CCND1–10, 13 and 20 (r2>0.8 for each pair of variants), with the minor alleles all-present in one haplotype (ATT), except for a single patient who carried the haplotype ATC. Therefore, these lower frequency variants were combined and analyzed as a single haplotype instead of independent variants. The aggregation of the CCND1–10/13/20 haplotype with the other nine variants with frequencies equal to or less than 5%, in the analysis, yielded lower ORs and a non-significant effect (Supplementary Table 4), as did a separate test on only the low frequency (1–5%) variants (Supplementary Table 5).
Functional analysis
We investigated the potential functional consequences of variants using in silico approaches. Promoter variants were examined for changes in transcription factor binding sites based on the matrices compiled by the program AliBaba 2.1. Several variants affect Sp1 sites (that is, CCND1–7, 10, 17), whereas CCND1–3 alters a CACCC box, which is recognized by transcription factors of the Sp/Krüppel-like Factor family. CCND1–13 creates a Cdx2 site. CCND1–18 has no apparent effect on transcription factor binding sites. For variants within the coding region, we examined the creation and/or elimination of alternative splicing sites. The rare C allele at CCND1–22 is predicted by the software tool ‘Human Splicing Finder’ to disrupt an acceptor site, generate a splicing enhancer site and also break up a splicing silencer site. In addition, it is well-known that the common variant rs9344 A allele modifies a splicing donor site and predisposes for an alternatively spliced transcript of the cyclin D1 protein (cyclin D1b).28 In transcript b, no splicing occurs at the exon 4-intron 4 boundary, and exon 5 is missing.1 Most variants found in 3′UTRs of the patients are reported to be within conserved blocks, as are the majority of the variants in the promoter regions and the exons. However, no 3′UTR variant altered a miRNA target site. There are no changes in the 5′UTR caused by CCND1–19 according to UTRdb, but ‘Human Splicing Finder’ indicates the potential creation of a splicing enhancer site (see Table 5 for a summary of the variants’ properties).
Because this functional analysis was done in silico, there remains the possibility that the predicted effects are not real and may need to be confirmed by direct experimental analysis.
rs9344 G/A and rare variants
The frequency of the rs9344 A allele was higher in UK and French cases as compared with controls, although not significantly so (Supplementary Table 3). French early-onset cases showed the highest frequency (0.53), which was, however, comparable to that reported for the CEU population. UK samples had lower frequencies than CEU, in agreement with findings for the British 1958 Birth Cohort (frequency of allele A=0.44).
Linkage disequilibrium analysis using Haploview indicates that rs9344 does not appear to be in strong LD with any rare or low frequency variant, and the correlation with the haplotype harboring the variants CCND1–10, 13 and 20, is also fairly weak (r2=0.02). However, examination of 2 × 2 tables reveals that LD between the CCND1–10/13/20 haplotype and rs9344 is nevertheless highly significant (Table 6). Given that it has been demonstrated that this polymorphism is not independently predictive of cancer and that additional events may be necessary to induce cyclin D1b production,28 we examined whether the presence of rare variants along the CCND1 gene is in any way associated with genotype at this position. We found that patients who carry at least one rare variant are more likely to also carry the rs9344 G allele than cases who do not harbor rare variants. A similar finding was obtained in controls although the effect was somewhat smaller (Supplementary Table 6). We believe, however, that this association is much more likely to be due to the effect of LD than to an actual relationship with disease.
Discussion
We have screened the coding and regulatory regions of the oncogene CCND1, a well-known candidate for colorectal cancer susceptibility, in about 150 UK individuals with multiple intestinal polyps or early-onset colorectal cancer. In total, 14 variants were selected for analysis, including nine rare variants (MAF<1%), four lower frequency variants (MAF, 1–5%) and one common variant (MAF>5%). Three of the rare variants had not been reported elsewhere (CCND1–17, 18, 22) and only CCND1–18, was detected in more than one affected subject. Three of the rare variants were not found in a set of over 800 UK controls (CCND1–19, 22, 28). Four variants were also typed in a set of French patients with similar disease phenotypes to those of the UK cases. One of these variants (CCND1–19) was not present in any of the French cases, whereas another one (CCND1–3) was present in French but not in UK patients. These results indicate that current catalogs of genetic variation are not exhaustive enough to represent low frequency variation among patients, which are also often likely to be population specific. However, more information on the genetic structure of the French patient sample is necessary, as well as a set of French controls, to extract significant conclusions about the impact of CCND1 rare variants on colorectal disease in this population.
Our findings show that, when all the rare variants are combined, there is a significant increase in the risk of colorectal disease with an OR of about 2. The effect appears to be stronger for developing multiple adenomas than for early-onset disease. In one case, CCND1–7, the effect was significant even for a single variant, with an OR greater than 3 for all UK cases.
When the data for all variants with frequencies lower than 5% (that is, rare and low frequency variants) were combined, the association was not significant. This suggests, as might be expected, that the lower the frequency of a disease-associated variant, the higher the OR is likely to be.
Nearly all the variants analyzed have putatively recognizable functional effects or lie within a conserved sequence block. However, no functional effects have yet been directly confirmed experimentally. CCND1–7 is a putative regulatory variant that alters two Sp1 binding sites. The Sp1 sites in the CCND1 promoter are highly conserved,29 and are required for the transcriptional activation of CCND1 following mitogenic stimulation.30, 31
It is interesting to note that, out of the 11 variants with MAF <5% identified in our study by chromatographic screening, 8 are located in non-coding regions of the gene, 6 of them in the 3′UTR. It has recently been established that genomic modifications of the CCND1 3′UTR in mantle cell lymphoma tumors produced mRNAs with truncated 3′UTRs that have considerably longer half lives than those of the full-length mRNAs.32 These transcripts are shorter versions of cyclin D1a and are not the alternatively spliced cyclin D1b isoform. Genetic changes that generate such transcripts include deletions of part of the 3′UTR or point mutations that create novel polyadenylation signals.32 Wiestner and colleagues believe that these alterations are somatic, although they did not examine germline DNA. Other authors33, 34 have also suggested the involvement of 3′UTR changes, including polymorphisms and rare deletions, as a cause of increased expression of CCND1 in cancer. In addition, loss of microRNA target sites as a result of 3′UTR shortening can lead to pathogenic overexpression of the protein.35 In our in silico analysis, however, none of the variants studied was suggested to modify a microRNA binding sequence.
We have also explored the putative relationship between a common variant that has been repeatedly associated with colorectal cancer, though inconsistently, and the presence of rare variants with potentially functional consequences. It could be the case that common and rare variants in the same gene or in different genes along the same pathway interact and modify each other's effects to produce the phenotype.19, 36
A search for possible combined effects of the common variant rs9344 and one or more of the rare or low-frequency variants is almost certainly likely to be confounded by weak LD between rs9344 and these variants, and so would require a much larger sample size. This is indicated by the very significant, but low level of LD between rs9344 and the CCND1–10/13/20 haplotype.
Our analysis of variation at the CCND1 gene has added to the evidence for the importance of rare variants as determinants of disease susceptibility. It has also shown how a moderate size study of rare variants in a candidate gene can reveal effects that are of clearly greater biological significance than very much larger whole-genome association studies (GWAS) of common variants. Thus, in these large case-control association studies only variants with frequencies higher than 5% are examined and the vast majority of significantly associated variants have been shown to give rise to very modest risk increases, generally with ORs not more than about 1.2. The case for rare variants has now been extensively discussed, both from observed data17, 18, 19, 37 and on the basis of theoretical considerations.38, 39, 40, 41 More recent studies have demonstrated additional rare variant influences on the pathogenesis of a variety of complex diseases and traits such as type 1 diabetes, colorectal cancer, plasma lipoprotein levels and neurological disorders.42, 43, 44, 45, 46, 47, 48, 49
Screening candidate genes in groups of patients for germline variation is the first step in unraveling rare variation, a step that is already being made much easier by the increasing accessibility of next-generation sequencing technologies. However, functional studies of the most interesting variants must follow closely. Cyclin D1 is a regulator of the entrance into the G1 phase of the cell cycle, and has been considered a promising predictive and prognostic biomarker for a number of cancers. Nevertheless, there have been few assessments of CCND1 levels of variation, with studies mostly focusing on the analysis of the functional polymorphism rs9344. More exhaustive studies that include rare and low frequency variants as well as evaluate regulatory regions are necessary if CCND1 is to be effectively used in the clinic.
References
Howe, D. & Lynas, C. The cyclin D1 alternative transcripts [a] and [b] are expressed in normal and malignant lymphocytes and their relative levels are influenced by the polymorphism at codon 241. Haematologica. 86, 563–569 (2001).
Arber, N., Hibshoosh, H., Moss, S. F., Sutter, T., Zhang, Y., Begg, M. et al. Increased expression of cyclin D1 is an early event in multistage colorectal carcinogenesis. Gastroenterology. 110, 669–674 (1996).
McKay, J. A., Douglas, J. J., Ross, V. G., Curran, S., Murray, G. I. & Cassidy, J. Cyclin D1 protein expression and gene polymorphism in colorectal cancer.. Int. J. Cancer. 88, 77–81 (2000).
Ashton, K. A., Proietto, A., Otton, G., Symonds, I., McEvoy, M., Attia, J. et al. The influence of the Cyclin D1 870 G>A polymorphism as an endometrial cancer risk factor. BMC Cancer. 8, 272 (2008).
Wilding, J., Straub, J., Bee, J., Churchman, M., Bodmer, W., Dickson, C. et al. Cyclin D1 is not an essential target of beta-catenin signaling during intestinal tumorigenesis, but it may act as a modifier of disease severity in multiple intestinal neoplasia (Min) mice. Cancer Res. 62, 4562–4565 (2002).
Pabalan, N., Bapat, B., Sung, L., Jarjanazi, H., Francisco-Pabalan, O. & Ozcelik, H. Cyclin D1 Pro241Pro (CCND1-G870A) polymorphism is associated with increased cancer risk in human populations: a meta-analysis. Cancer Epidemiol. Biomarkers Prev. 17, 2773–2781 (2008).
Kong, S., Amos, C. I., Luthra, R., Lynch, P. M., Levin, B. & Frazier, M. L. Effects of cyclin D1 polymorphism on age of onset of hereditary nonpolyposis colorectal cancer. Cancer Res. 60, 249–252 (2000).
Talseth, B. A., Ashton, K. A., Meldrum, C., Suchy, J., Kurzawski, G., Lubinski, J. et al. Aurora-A and cyclin D1 polymorphisms and the age of onset of colorectal cancer in hereditary nonpolyposis colorectal cancer. Int. J. Cancer. 122, 1273–1277 (2008).
Chen, J., Etzel, C. J., Amos, C. I., Zhang, Q., Viscofsky, N., Lindor, N. M. et al. Genetic variants in the cell cycle control pathways contribute to early onset colorectal cancer in Lynch syndrome. Cancer Causes Control. 20, 1769–1777 (2009).
Kong, S., Wei, Q., Amos, C. I., Lynch, P. M., Levin, B., Zong, J. et al. Cyclin D1 polymorphism increased risk of colorectal cancer at young age. J. Natl. Cancer Inst. 93, 1106–1108 (2001).
Porter, T. R., Richards, F. M., Houlston, R. S., Evans, D. G. R., Jankowski, J. A., Macdonald, F. et al. Contribution of cyclin d1 (CCND1) and E-cadherin (CDH1) polymorphisms to familial and sporadic colorectal cancer. Oncogene. 21, 1928–1933 (2002).
Grünhage, F., Jungck, M., Lamberti, C., Berg, C., Becker, U., Schulte-Witte, H. et al. Association of familial colorectal cancer with variants in the E-cadherin (CDH1) and cyclin D1 (CCND1) genes. Int. J. Colorectal Dis. 23, 147–154 (2008).
Schernhammer, E. S., Tranah, G. J., Giovannucci, E., Chan, A. T., Ma, J., Colditz, G. A. et al. Cyclin D1 A870G polymorphism and the risk of colorectal cancer and adenoma. Br. J. Cancer. 94, 928–934 (2006).
Tan, X.- L., Nieters, A., Kropp, S., Hoffmeister, M., Brenner, H. & Chang-Claude, J. The association of cyclin D1 G870A and E-cadherin C-160A polymorphisms with the risk of colorectal cancer in a case control study and meta-analysis. Int. J. Cancer. 122, 2573–2580 (2008).
Bala, S. & Peltomaki, P. Cyclin D1 as a genetic modifier in hereditary nonpolyposis colorectal cancer. Cancer Res. 61, 6042–6045 (2001).
Kruger, S., Engel, C., Bier, A., Mangold, E., Pagenstecher, C., Von Knebel Doeberitz, M. et al. Absence of association between cyclin D1 (CCND1) G870A polymorphism and age of onset in hereditary nonpolyposis colorectal cancer. Cancer Lett. 236, 191–197 (2006).
Bodmer, W. Familial adenomatous polyposis (FAP) and its gene, APC. Cytogenet. Genome Res. 86, 99–104 (1999).
Fearnhead, N. S., Wilding, J. L., Winney, B., Tonks, S., Bartlett, S., Bicknell, D. C. et al. Multiple rare variants in different genes account for multifactorial inherited susceptibility to colorectal adenomas. Proc. Natl Acad. Sci. USA. 101, 15992–15997 (2004).
Bodmer, W. & Bonilla, C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 40, 695–701 (2008).
Frayling, I. M., Beck, N. E., Ilyas, M., Dove-Edwin, I., Goodman, P., Pack, K. et al. The APC variants I1307 K and E1317Q are associated with colorectal tumors, but not always with a family history. Proc. Natl Acad. Sci. USA. 95, 10722–10727 (1998).
Lamlum, H., Al Tassan, N., Jaeger, E., Frayling, I., Sieber, O., Reza, F. B. et al. Germline APC variants in patients with multiple colorectal adenomas, with evidence for the particular importance of E1317Q. Hum. Mol. Genet. 9, 2215–2222 (2000).
Bodmer, W. F. & Bodmer, J. G. Cytofluorochromasia for HLA A, B, C and DR typing. In: NIAID Manual of Tissue Typing Techniques 1979–1980, Vol. 80. NIH Publication: Bethesda, MD, pp 545–546.
Ahn, S. J., Costa, J. & Rettig Emanuel, J. PicoGreen quantitation of DNA: effective evaluation of samples pre- or post-PCR. Nucleic Acids Res. 24, 2623–2625 (1996).
Liu, K. & Muse, S. V. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 21, 2128–2129 (2005).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 21, 263–265 (2005).
Desmet, F.- O., Hamroun, D., Lalande, M., Collod-Béroud, G., Claustres, M. & Béroud, C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67 (2009).
Coassin, S., Brandstätter, A. & Kronenberg, F. Lost in the space of bioinformatic tools: a constantly updated survival guide for genetic epidemiology. The GenEpi Toolbox. Atherosclerosis. 209, 321–335 (2010).
Comstock, C. E. S., Augello, M. A., Benito, R. P., Karch, J., Tran, T. H., Utama, F. E. et al. Cyclin D1 splice variants: polymorphism, risk, and isoform-specific regulation in prostate cancer. Clin. Cancer Res. 15, 5338–5349 (2009).
Eto, I. Molecular cloning and sequence analysis of the promoter region of mouse cyclin D1 gene: implication in phorbol ester-induced tumour promotion. Cell Prolif. 33, 167–187 (2000).
Nagata, D., Suzuki, E., Nishimatsu, H., Satonaka, H., Goto, A., Omata, M. et al. Transcriptional activation of the cyclin D1 gene is mediated by multiple cis-elements, including SP1 sites and a cAMP-responsive element in vascular endothelial cells. J. Biol. Chem. 276, 662–669 (2001).
Bartusel, T., Schubert, S. & Klempnauer, K.- H. Regulation of the cyclin D1 and cyclin A1 promoters by B-Myb is mediated by Sp1 binding sites. Gene. 351, 171–180 (2005).
Wiestner, A., Tehrani, M., Chiorazzi, M., Wright, G., Gibellini, F., Nakayama, K. et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood. 109, 4599–4606 (2007).
Sathyan, K. M., Nalinakumari, K. R., Abraham, T. & Kannan, S. CCND1 polymorphisms (A870G and C1722G) modulate its protein expression and survival in oral carcinoma. Oral Oncol. 44, 689–697 (2008).
Hosokawa, Y., Suzuki, R., Joh, T., Maeda, Y., Nakamura, S., Kodera, Y. et al. A small deletion in the 3′-untranslated region of the cyclin D1/PRAD1/bcl-1 oncogene in a patient with chronic lymphocytic leukemia. Int. J. Cancer. 76, 791–796 (1998).
Deshpande, A., Pastore, A., Deshpande, A. J., Zimmermann, Y., Hutter, G., Weinkauf, M. et al. 3′UTR mediated regulation of the cyclin D1 proto-oncogene. Cell Cycle. 8, 3584–3592 (2009).
Felix, R., Bodmer, W., Fearnhead, N. S., van der Merwe, L., Goldberg, P. & Ramesar, R. S. GSTM1 and GSTT1 polymorphisms as modifiers of age at diagnosis of hereditary nonpolyposis colorectal cancer (HNPCC) in a homogeneous cohort of individuals carrying a single predisposing mutation. Mut. Res. 602, 175–181 (2006).
Cohen, J. C., Kiss, R. S., Pertsemlidis, A., Marcel, Y. L., McPherson, R. & Hobbs, H. H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 305, 869–872 (2004).
Pritchard, J. K. & Cox, N. J. The allelic architecture of human disease genes: common disease-common variant.or not? Hum. Mol. Genet. 11, 2417–2423 (2002).
Schork, N. J., Murray, S. S., Frazer, K. A. & Topol, E. J. Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 19, 212–219 (2009).
Goldstein, D. B. Common genetic variation and human traits. New Eng. J. Med. 360, 1696–1698 (2009).
Eyre-Walker, A. Evolution in health and medicine Sackler colloquium: genetic architecture of a complex trait and its implications for fitness and genome-wide association studies. Proc. Natl Acad. Sci. USA. 107, 1752–1756 (2010).
Nejentsev, S., Walker, N., Riches, D., Egholm, M. & Todd, J. A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 324, 387–389 (2009).
Kaufmann, A., Vogt, S., Uhlhaas, S., Stienen, D., Kurth, I., Hameister, H. et al. Analysis of rare APC variants at the mRNA level: six pathogenic mutations and literature review. J. Mol. Diagn. 11, 131–139 (2009).
Azzopardi, D., Dallosso, A. R., Eliason, K., Hendrickson, B. C., Jones, N., Rawstorne, E. et al. Multiple rare nonsynonymous variants in the adenomatous polyposis coli gene predispose to colorectal adenomas. Cancer Res. 68, 358–363 (2008).
Mefford, H. C., Cooper, G. M., Zerr, T., Smith, J. D., Baker, C., Shafer, N. et al. A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease. Genome Res. 19, 1579–1585 (2009).
Burnett, J. R. & Hooper, A. J. Common and rare gene variants affecting plasma LDL cholesterol. Clin. Biochem. Rev. 29, 11–26 (2008).
Cohen, J. C., Pertsemlidis, A., Fahmi, S., Esmail, S., Vega, G. L., Grundy, S. M. et al. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc. Natl Acad. Sci. USA. 103, 1810–1815 (2006).
Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M. et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 320, 539–543 (2008).
Bearden, C. E., Glahn, D. C., Lee, A. D., Chiang, M.- C., Van Erp, T. G. M., Cannon, T. D. et al. Neural phenotypes of common and rare genetic variants. Biol. Psychol. 79, 43–57 (2008).
Acknowledgements
We would like to thank all individuals who contributed samples for this study. We are also grateful to Shazad Asraf, Oliver Sieber, and Haitao Wang for making samples available, and to Jennifer Wilding for helpful comments. We thank the patients who participated in VICTOR, without whose help the study would not have been possible. The trial was enabled by an educational study grant from Merck, which also supplied the study drug. VICTOR was endorsed by the UK National Cancer Research Institute (NCRI) and Cancer Research Campaign Clinical Trials Awards and Advisory Committee (CTAAC), allowing support from the UK National Cancer Research Network. The University of Birmingham was the centre for randomization and for statistics initially before these moved to the Universities of Oxford and Warwick, respectively. We also thank VICTOR trial staff: A Stanley, J Stokes, F Duchesne, K Reed, S Pendlebury, R Duvvuri, I Kennedy, G Davis, J Birtwistle, L Blair, A Bange, K Bell, L Dunham, R Deacon, A Mellor, B Mikolajczyk, M Davoudianfar and A Ferch; and VICTOR laboratory support staff: K Elliot, M Presz, H Wang; Clinical Coordinator: D Rea; The VICTOR Trial Advisory Group and the VICTOR Data and Safety Monitoring Committee, and all nurses and medical staff who participated in this trial. CB was Cancer Research UK Julia Bodmer Fellow. This work was made possible by a Cancer Research UK program grant, a Wellcome Trust grant and a Serrousi Foundation grant to WFB.
Author information
Authors and Affiliations
Corresponding author
Additional information
URLs
AliBaba 2.1 http://www.gene-regulation.com/pub/programs/alibaba2/index.html
British Birth Cohort http://www.b58cgene.sgul.ac.uk/index.php
dbSNP http://www.ncbi.nlm.nih.gov/projects/SNP/
GenEpi toolbox http://genepi_toolbox.i-med.ac.at/
Human Splicing Finder (HSF) http://www.umd.be/HSF/
People of the British Isles study (PoBI) http://www.peopleofthebritishisles.org
UTRdb http://utrdb.ba.itb.cnr.it/
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Bonilla, C., Lefèvre, J., Winney, B. et al. Cyclin D1 rare variants in UK multiple adenoma and early-onset colorectal cancer patients. J Hum Genet 56, 58–63 (2011). https://doi.org/10.1038/jhg.2010.144
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.144
Keywords
This article is cited by
-
Cyclin D1 protein affecting global women’s health by regulating HPV mediated adenocarcinoma of the uterine cervix
Scientific Reports (2019)
-
Colorectal Cancer in the Young
Current Gastroenterology Reports (2018)
-
Role of rare variants in undetermined multiple adenomatous polyposis and early-onset colorectal cancer
Journal of Human Genetics (2012)
