
Abstract
Mutations in the lamin A/C gene (LMNA) are established causes of familial dilated cardiomyopathy (DCM) with atrio-ventricular block although relatively little is known about genotype–phenotype correlations. We describe a 23-year-old patient who presented with inferolateral wall thinning and akinesis with evidence of mid-myocardial fibrosis on cardiac magnetic resonance. Molecular analysis driven by clinical similarities with a previously described case harboring the p.R541C LMNA mutation revealed a novel c.1621 C>G, p.R541G substitution whose pathogenicity was confirmed by transfection of mouse myoblasts. Our results emphasize the role of LMNA mutations at position R541 in DCM cases with segmental LV wall motion akinesis/dyskinesis.
Similar content being viewed by others

Introduction
Mutations in the lamin A/C gene (LMNA, Genbank accession number: L123399, L12400 and L12401) are responsible for a significant proportion of cases with familial dilated cardiomyopathy (DCM).1, 2, 3 In these cases, DCM is usually preceded by arrhythmias and conduction abnormalities, and often leads to severe heart failure4, 5 although reliable genotype–phenotype correlations are difficult to delineate. Cardiac magnetic resonance (CMR) can help elucidate discrete dysfunctional areas of the left ventricle (LV) and demonstrate a characteristic pattern of non-ischaemic delayed enhancement, signifying fibrosis.6, 7, 8 We describe a patient from a previously investigated family9 with ventricular arrhythmias preceding DCM and evidence of apical aneurysm in his aunt. The patient presented with regional wall motion abnormalities on CMR and was found to have a novel functional LMNA mutation. Interestingly, the molecular diagnosis was driven by phenotypic similarities with a previosuly described unrelated case of DCM harboring the p.R541C LMNA mutation.5
Case report
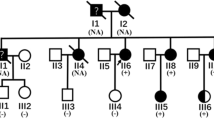
A 23-year-old man with a family history of DCM was referred to us for the evaluation of asymptomatic LV dilation diagnosed 3 years earlier. The father of the patient had an angiographically proven DCM (percentage left ventricular enlargement=162, left ventricular ejection fraction [LVEF]=23%), severe ventricular arrhythmias and died while on the waiting list for heart transplantation at the age of 30 years (Figure 1). The father's sister was diagnosed at the age of 31 years and had global hypokinesis of the LV with mildly reduced LVEF, apical LV aneurysm and complex ventricular arrhythmias. Coronary angiography was normal. She died suddenly at the age of 39 years.

Family pedigrees. The arrow shows the proband. Blackened squares (male) and circles (female) indicate confirmed cases of DCM. Open symbols represent subjects not examined. N indicates phenotypically normal subjects. Shaded symbol indicates a likely obligate carrier. Stars indicate genotyped patients. Roman numerals indicate generations. Numbers above symbols indicate consecutive examined carriers.
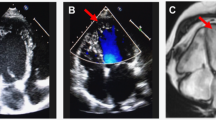
Our patient (proband) was free from heart failure symptoms (NYHA class I), but underwent radiofrequency ablation for paroxysmal atrio-ventricular nodal re-entrant tachycardia 6 years before the current presentation. ECG demonstrated sinus rhythm with nonspecific intraventricular block and QRS of 124 ms and presence of Q waves in leads I, aVL and V1-V2 (Figure 2a). Transthoracic echocardiography revealed dilation of the LV (percentage left ventricular enlargement=121) and the presence of mild global hypokinesis (LVEF=50%). The patient received angiotensin converting enzyme inhibitor and beta-blocker. To further characterize the LV morphology and function, the patient was referred for CMR. The study confirmed markedly increased LV end-diastolic volume with decreased LVEF=44% and demonstrated discrete regional areas of akinesis with muscle thinning (5 mm) located in the mid-ventricular and periapical inferolateral segments of the LV (Figures 2b and c). A marked hypertrabeculation in dysfunctional regions was also seen. Scans performed 10–15 min after gadolinium contrast injection revealed mid-myocardial, almost transmural areas of delayed enhancement in the dysfunctional segment (Figures 2d and e). To exclude the ischemic origin of the LV dysfunction, the patient was subjected to coronary CT angiography which did not disclose any changes in coronary arteries (not shown).

Clinical findings in the studied patient: (a) 12-lead electrocardiogram; (b–e) Cardiac magnetic resonance: (b, c) marked thinning of the mid-ventricular inferolateral segment in cine SSFP images in end-diastole (arrow); (d, e) the presence of mid-myocardial almost transmural fibrosis in the same segments in phase sensitive inversion recovery (PSIR) images with visible delayed enhancement (arrows). RV, right ventricle, LV, left ventricle.
Laboratory findings included mildly increased troponin levels, with NT-proBNP concentrations within reference range. Cardiopulmonary treadmill test showed generally preserved physical capacity (V02max=29.6 ml kg−1 min−1, 62% predicted at RER 1.08, 8.5 METs) without complex ventricular extrasystole at peak exercise. Ambulatory ECG monitoring on carvedilol (25 mg per day) demonstrated 196 premature ventricular beats without complex forms of arrhythmia. Patient was scheduled to undergo ambulatory ECG monitoring every 3 months with subsequent restratification for ICD implantation at 6–12 months time.
Because of the family history (apical LV aneurysm in the patient's aunt, ventricular arrhythmias in the patient's aunt and father), presence of the intraventricular block and CMR evidence of regional LV akinesis, we decided to screen the patient for LMNA mutations as described previously.5 Assumption of possible LMNA mutation was based on similar phenotype reported in patients with p.R541C LMNA mutation initially described by Forrisier et al.4 (LV apical aneurysm, severe ventricular rhythm disturbances, left bundle branch block) and later by Hookana et al.7 (akinesis of the posterior LV wall, severe ventricular arrhythmias and sudden cardiac death) and by our group5 (dyskinesis of the inferior LV wall, akinesis of the LV apex, severe ventricular arrhythmias and left bundle branch block). Analysis excluded presence of p.R541C but, interestingly revealed a previously undescribed mutation at the same position, with arginine to glycine substitution (c.1621 C>G, p.R541G) (Figure 3a). Cellular transfection of fluorescently labeled lamin A/C with c.1621 C>G, p.R541G led to the development of abnormal lamin aggregates in about 80% of the cells (Figure 3b) compared with homogeneous nuclear distribution in wild type (Figure 3d). In 60% of these aggregated cells, the aggregates formed sickle-shaped aggregates (Figure 3c) as opposed to the common circular aggregates.10

(a) Result of direct sequencing demonstrating Arginine to Glycine (c.1621 C>G, p.R541G) substitution in exon 10 of LMNA. (wt, normal variant; mut, variant with mutation). (b–d) C2C12 mouse myoblasts transiently transfected with c.1621 C>G, p.R541G mutant lamin A and lamin C in fluorescent expression vectors as described previously:10 (b) example of a cell with aggregates, (c) example of a cell with sickle-shaped aggregates—see text for details. For comparison (d) the wild type of lamins A and C transfected together with homogeneous distribution of the protein throughout the nucleus.
The same mutation was found in the patient's sister, age 28, who had a sinus bradycardia on ECG with the presence of Q waves in leads II and V4-V6. She suffered from palpitations, but was free from symptoms of heart failure. Holter monitoring revealed supraventricular and ventricular arrhythmias (pairs, periods of bigeminia). She had a mildly elevated troponin levels, but normal echocardiographic (percentage left ventricular enlargement=100) and CMR picture.
Discussion
We present a patient belonging to a second generation of the family with confirmed DCM who was found to have a novel functional c.1621 C>G, p.R541G LMNA mutation.
The R541 site is located in the C-terminal tail region of the lamin A/C protein. The tail apears to promote secondary structural organization of lamin dimers in the formation of longitudinal polar head-to-tail polymers.10, 11 The novel point mutation results in the change of the arginine into a glycine, which, contrary to arginine, has a nonpolar neutral side chain. Experiments in cellular models demonstrated that c.1621 C>G, p.R541G mutation presents a phenotype with multiple small aggregates of the mutant proteins, and frequent formation of sickles connecting the mutant aggregates (Figures 3b–d). Cellular expression of DCM-associated p.R541S presented with no aberrant phenotype.10 The formation of intranuclear aggregates is commonly observed in the presence of disease-associated LMNA mutations;10, 12, 13, 14 however, the presence of sickles has yet to be reported in disease-associated mutants. This indicates aberrant formation of the inner nuclear lamina, most likely because of misassembled lamin dimers caused by polarity change.
The phenotype of the novel mutation includes electrocardiographic abnormalities (nonspecific intraventricular block and pathological Q waves), regional LV akinesis/dyskinesis (found in the patient and his aunt) and ventricular arrhythmias (found in the patient, patient's father, aunt and sister). These manifestations are very similar to those observed in patients harboring another substitution at the same position which replaces arginine with cysteine.4, 5, 7 However, in addition to the exact location of the mutation, expression of this phenotype clearly depends on the specific amino acid change as evidenced by reports of p.R541S mutation10 and p.R541H mutation,15 both of which had no evidence of regional wall motion anomalies. It should be also mentioned that, in contrast to p.R541H mutation, in our patients there were no signs or symptoms of muscular dystrophy.15 In this context, it is relevant to note that glycine and cysteine (but not serine and histidine) both have nonpolar side chains and, thus, may have a similar functional effect.
Regional wall motion abnormalities (mostly various degrees of hypokinesis) may be present even in 50% of patients with DCM.16 However, the presence of regional dyskinesis is a rare finding among DCM patients mainly found in subjects after previous myocarditis.17 CMR is a sensitive tool in the diagnosis of discrete segmental LV dysfunction, which may be easily overlooked in routine transthoracic echocardiography, especially when it is localized in the posterior wall. CMR examination can demonstrate the characteristic mid-wall pattern of delayed enhancement (fibrosis) in the dysfunctional regions6, 7 and the presence of hypertrabeculation1 which is also associated with LMNA mutations. Nevertheless, it should be mentioned that CMR may not be able to demonstrate early signs of DCM even in patients with abnormal ECG pattern, ventricular arrhythmia and elevated troponin levels as demonstrated in the patient's sister. The lack of visible abnormalities on CMR examination in this case may be at least partially related to later onset of the disease in women in comparison to men,9, 18 as observed in the patient's aunt. Therefore, the importance of genetic counseling regarding risk of sudden cardiac death and heart failure in families harboring LMNA mutations should be emphasized.
References
Elliott, P., Andersson, B., Arbustini, E., Bilinska, Z., Cecchi, F., Charron, P. et al. Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur. Heart. J. 29, 270–276 (2008).
Møller, D. V., Pham, T. T., Gustafsson, F., Hedley, P., Ersbøll, M. K., Bundgaard, H. et al. The role of lamin A/C mutations in Danish patients with idiopathic dilated cardiomyopathy. Eur. J. Heart. Fail. 11, 1031–1035 (2009).
Sylvius, N. & Tesson, F. Lamin A/C and cardiac diseases. Curr. Opin. Cardiol. 21, 159–165 (2006).
Forissier, J. F., Bonne, G., Bouchier, C., Duboscq-Bidot, L., Richard, P., Wisnewski, C. et al. Apical left ventricular aneurysm without atrio-ventricular block due to a lamin A/C gene mutation. Eur. J. Heart. Fail. 5, 821–825 (2003).
Saj, M., Jankowska, A., Lewandowski, M., Szwed, H., Szperl, M., Ploski, R. et al. Dilated cardiomyopathy with profound segmental wall motion abnormalities and ventricular arrhythmia caused by the R541C mutation in the LMNA gene. Int. J. Cardiol. 144, e51–e53 (2009).
Raman, S. V., Sparks, E. A., Baker, P. M., McCarthy, B. & Wooley, C. F. Mid-myocardial fibrosis by cardiac magnetic resonance in patients with lamin A/C cardiomyopathy: possible substrate for diastolic dysfunction. J. Cardiovasc. Magn. Reson. 9, 907–913 (2007).
Hookana, E., Juntilla, M. J., Särkioja, T., Sormunen, R., Niemelä, M., Raatikainen, M. J. et al. Cardiac arrest and left ventricular fibrosis in a Finish family with the lamin A/C mutation. J. Cardiovasc. Electrophysiol. 19, 743–747 (2008).
Koikkalainen, J. R., Antila, M., Lötjönen, J. M., Heliö, T., Lauerma, K., Kivistö, S. M. et al. Early familial dilated cardiomyopathy: identification with determination of disease state parameter from cine MR image data. Radiology 249, 88–96 (2009).
Bilinska, Z. T., Michalak, E., Piatosa, B., Grzybowski, J., Skwarek, M., Deptuch, T. W. et al. Familial dilated cardiomyopathy: evidence for clinical and immunogenetic heterogeneity. Med. Sci. Monit. 9, CR219–226 (2003).
Sylvius, N., Bilinska, Z. T., Veinot, J. P., Fidzianska, A., Bolongo, P. M., Poon, S. et al. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J. Med. Genet. 42, 639–647 (2005).
Sasse, B., Aebi, U. & Stuurman, N. A tailless Drosophila lamin Dm0 fragment reveals lateral associations of dimers. J. Struct. Biol. 123, 56–66 (1998).
Raharjo, W. H., Enarson, P., Sullivan, T., Stewart, C. L. & Burke, B. Nuclear envelope defects associated with LMNA mutations cause dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy. J. Cell. Sci. 114, 4447–4457 (2001).
Ostlund, C., Bonne, G., Schwartz, K. & Worman, H. J. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J. Cell. Sci. 114, 4435–4445 (2001).
Sylvius, N., Hathaway, A., Boudreau, E., Gupta, P., Labib, S., Bolongo, P. M. et al. Specific contribution of lamin A and lamin C in the development of laminopathies. Exp. Cell. Res. 314, 2362–2375 (2008).
Vytopil, M., Benedetti, S., Ricci, E., Galluzzi, G., Dello Russo, A., Merlini, L. et al. Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J. Med. Genet. 40, e132 (2003).
Yamaguchi, S., Tsuiki, K., Hayasaka, M. & Yasui, S. Segmental wall motion abnormalities in dilated cardiomyopathy: hemodynamic charcteristics and comparison with thallium-201 myocardial scintigraphy. Am. Heart J. 113, 1123–1128 (1987).
Chimenti, C., Calabrese, F., Thiene, G., Pieroni, M., Maseri, A. & Frustaci, A. Inflammatory left ventricular microaneurysms as a cause of apparently idiopathic ventricular tachyarrhythmias. Circulation 104, 168–173 (2001).
Mangin, L., Charron, P., Tesson, F., Mallet, A., Dubourg, O., Desnos, M. et al. Familial dilated cardiomyopathy: clinical features in French families. Eur. J. Heart. Fail. 1, 353–361 (1999).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Małek, Ł., Labib, S., Mazurkiewicz, Ł. et al. A new c.1621 C>G, p.R541G lamin A/C mutation in a family with DCM and regional wall motion abnormalities (akinesis/dyskinesis): genotype–phenotype correlation. J Hum Genet 56, 83–86 (2011). https://doi.org/10.1038/jhg.2010.137
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.137
Keywords
This article is cited by
-
LMNA mutations in Polish patients with dilated cardiomyopathy: prevalence, clinical characteristics, and in vitro studies
BMC Medical Genetics (2013)
-
Variants of the Lamin A/C (LMNA) Gene in Non-Valvular Atrial Fibrillation Patients
Molecular Diagnosis & Therapy (2012)
