
Abstract
The Khon Mueang (KM) are the largest group of northern Thai people. Our previous mtDNA studies have suggested an admixture process among the KM with the earlier Mon-Khmer-speaking inhabitants of this region. In this study, we evaluate genetic affinities and admixture among 10 KM populations in northern Thailand lying along the historical Yuan migration route, and 10 neighboring populations belonging to 7 additional ethnic groups: Lawa, Mon (Mon-Khmer-speaking groups), Shan, Yuan, Lue, Khuen and Yong (Tai-speaking groups) by analyzing 15 hypervariable autosomal short tandem repeat loci. The KM exhibited close relationships with neighboring populations, especially the Tai-speaking groups, reflecting an admixed origin of the KM. Admixture proportions were observed in all KM populations, which had a higher contribution from the parental Tai than the Mon-Khmer groups. Different admixture patterns of the KM along the migration route might indicate high heterogeneity among the KM. These patterns were not directly associated with geographical proximity, suggesting other factors, like variation in the timing of admixture with the existing populations may have had an important role. More genetic data from different marker systems solely transmitted through the male or female lineages are needed to complete the description of genetic admixture and population history of the KM.
Similar content being viewed by others


Introduction
Northern Thailand is in the landlocked heart of the golden peninsula of southeast Asia, characterized river plains, separated by steep mountainous ranges. It shares a border with Myanmar and Laos, and lies in close proximity to southern China. This territory was primarily inhabited by the indigenous Lawa (LW), who probably descended from younger prehistoric peoples around the fifth century AD.1, 2, 3, 4, 5 Subsequently, the area became ethnically, culturally and linguistically diversified through migration and settlement of the Mon and several Tai-speaking populations. The MO (MO) were among the oldest settlers in Thailand and established an advanced civilization in central Thailand around the fifth century AD. During the second half of the eighth century AD, they extended their kingdom as far as today's Lamphun province in northern Thailand.1
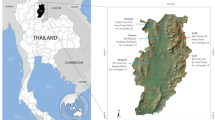
The Yuan (YU), whose origins remain mysterious, are regarded as the first Tai-speaking population to reach Thailand. By the eight century, they had established the first Tai kingdom, Chiang Sean, located in present-day Chiang Rai province.2 By the end of thirteenth century AD, the Yuan migration began with the invasion of Mon-speaking lands in Lamphun. They founded a new city at Chiang Mai and then extended their kingdom, named Lan Na, southward to Lampang.1, 2, 3, 4, 5 Apart from the Mon-Khmer and the Yuan groups, residing along the historical Yuan migratory route (Figure 1), this area was also populated by other Tai peoples: Lue, Yong, Khuen and Shan.

Geographical distribution of studied populations. See the meaning of population abbreviations in Table 1. The migration route of Khon Mueang is represented by dash arrow.
The Lue (LU) kingdom named Chiang Hung was founded in southern China around the twelfth century. At the present time, the Lue are distributed over parts of southern China, northern Myanmar, northern Laos and northern Thailand.2, 6 People from two Lue villages (LU1, LU2) in this study are thought to have migrated from different cities in Xishuangbanna through Laos into northern Thailand about 200–300 years ago.7 The Yong (YO) were residents of Yong city (Mong Yawng) in Shan State of Myanmar, and were probably originally Lue. They were taken captive by war, and forcibly migrated into Thailand in 1805 AD.4, 7 The city of the Khuen (KH), located at Keng Tung in the Shan State of Myanmar, was built by the Yuan king in 1267 AD.4 Their migration into Thailand was part of the same set of events that displaced the Yong. The Lue, Yong and Khuen's colloquialisms, script and their spoken dialects are similar to each other.7 The Shan (SH) or Tai Yai, which means great Tai, predominantly reside in the Shan State of Myanmar that bears their name. After their many migrations into northern Thailand, beginning around 1400 AD, the Shan today reside in the mountainous areas of Mae Hong Son province. With each successive generation, some of these people retained their original ethnicity, while those who assimilated became the present-day Khon Mueang (KM).2
Among the people living in present day northern Thailand, the Khon Mueang, speaking a Tai-Kadai language1 comprise the majority. ‘Khon Mueang’ is the local name commonly applied by the people of northern Thailand to themselves,8, 9 but this word now applies mostly to a social and political category rather than a distinct population, and it is not the ethnic name of any specific group. Nevertheless, the biological ancestry of the Khon Mueang has been variously proposed to originate from local Mon-Khmer-speaking peoples; the Lawa and Mon, assimilated by the Tai rulers; or via direct descent from the Tai-speaking groups who had migrated from southern China.3, 10, 11 Our previous analysis of mtDNA variation in the Khon Mueang and other Tai-speaking groups supported this latter hypothesis: that the Khon Mueang ancestors originated from Tai-speaking groups in south and southeast China, with subsequent admixture with Mon-Khmer populations.7
Admixture, the genetic consequence of mixing between genetically differentiated populations, resulting from geographical, ecological or cultural separation, is common in human evolution. Pioneering research on genetic admixture of human populations was mainly been applied to African-Americans,12, 13, 14, 15 Latin-Americans,16 Europeans17, 18 and East Asians,19, 20, 21 but has been limited in Southeast Asia.
Geography had an important role in the location of Tai settlements. River plains, surrounded by mountain peaks, were chosen because of their high agricultural productivity. Three geographically separated basins; the Chiang Rai Basin in the Kok River valley, the Chiang Mai-Lamphun Basin in the Ping River valley and the Lampang Basin in the Wang River valley, were the most suitable areas, along their migration route from Chiang Rai to Lampang province (Figure 1).4
Microsatellites, also known as short tandem repeats (STRs) have been broadly used for inferring human population history22, 23 and relationships among continental populations24 as well as between geographically contiguous populations.25, 26, 27 In this research work, autosomal STR variation analysis of 10 present-day Khon Mueang villages from the areas of Yuan settlement, as well as 10 other populations, who had an important role in northern Thai history (and, hence, could potentially be parental populations of the Khon Mueang) were performed, to investigate the population affinity of the studied populations and to explore the level of admixture in the Khon Mueang.
Materials and methods
Samples
Blood samples were collected from 943 unrelated healthy individuals (436 from Khon Mueang and 507 from neighboring populations), with informed consent, after interviewing them about their linguistic, village and personal history. Ten Khon Mueang (KM) populations reside along the Yuan migratory route in three different basins. KM1 reside in the Chiang Rai Basin, whereas KM2–KM6 and KM8 are in the Chiang Mai-Lamphun Basin, except KM7, who reside in the Mae Cham Basin of Chiang Mai (which is not on the Yuan migration route). KM9 and KM10 settled in the Lampang Basin (Figure 1). Based on historical sources, 10 neighboring populations, belonging to 7 ethnic groups, were also sampled: the Lawa (LW1, LW2), Mon (MO), Shan (SH), Yuan (YU1, YU2), Khuen (KH), Lue (LU1, LU2) and Yong (YO), who have had historical contact with the KM. DNA samples of the YU, KH, LU and YO from the previous study7 were also included in this study. General information about the studied populations is listed in Table 1.
DNA isolation and STR typing
Genomic DNA was extracted, using a standard inorganic salting-out method.28 Multiplex PCR amplification was performed, using a commercial AmpFISTR Identifiler kit (Applied Biosystem, Foster City, CA, USA), including primers for 15 autosomal STR loci: D8S1179, D21S11, D7S820, CSF1PO, D3S1358, THO1, D13S317, D16S539, vWA, TPOX, D18S51, D5S818, FGA, D19S433 and D2S1338, by reducing the total reaction volume to 5 μl. Amplicons were separated by multicapillary electrophoresis in an ABI 3100 genetic analyzer (Applied Biosystem). Results were then analyzed by GeneMapper software v.3.7 (Applied Biosystem).
Statistical analyses
STR allele frequencies, Hardy–Weinberg P-values, observed and expected heterozygosity (HO and HE, respectively), total allele and gene diversity indices were calculated with ARLEQUIN 3.1.29 The observed heterozygosity, averaged within the populations, was tested for heterogeneity with the Kruskal–Wallis test,30 using the commercially available STATISTICA 7.0 software package (StatSoft, Padova, Italy). The relative amount of gene flow into each population was calculated according to the model of Harpending and Ward (1982),31 which uses the theoretic regression of population heterozygosity and distance to the centroid. As these markers are widely used for forensic study, several parameters of forensic and population genetic importance including power of discrimination, matching probability, polymorphic information content, power of exclusion and typical paternity index were obtained with the Excel PowerStats spread sheet (www.promega.com/geneticidtools/powerstats).
To clarify population affinity, pair-wise genetic distances based on allele frequency variance (Fst), were computed by ARLEQUIN 3.1. The distance matrix was then plotted in two dimensions, by means of multidimensional scaling (MDS), using the STATISTICA 7.0 software.
The correlation among genetic and geographical distances was assessed by the Mantel test32 employing ARLEQUIN 3.1. Geographical distances between the approximate locations of each population were computed as great-circle distances calculated from their latitudinal and longitudinal coordinates.
One of the most important factors in admixture analysis is the level of divergence between the parental populations,33 thus, both distance-based and model-based approaches were employed to investigate genetic differentiation among the potential parental populations. Pair-wise genetic distances based on allele frequency variances (Fst) were computed using ARLEQUIN 3.1. Significance levels for these values were adjusted by sequential Bonferroni correction.34 Population structure was also investigated by means of the Bayesian clustering method implemented in STRUCTURE 2.335, 36, 37 under assumptions of admixture, correlated allele frequencies and either with or without (LOCPRIOR model37) assistance of sampling location as prior information. When the LOCPRIOR model was used, the value of the parameter r was also reported. An r value lower than 1, indicates that sampling location in the model is effective.37 For each number of clusters (K) from 1 to 10, five runs were performed, using an MCMC chain burn-in length of 100 000 iterations followed by 1 000 000 iterations. Chain convergence was assessed by comparing results from five different chains. For each K, the posterior probability of clustering was estimated from the average logarithmic probability of data across runs. Finally, second order rate of change of logarithmic probability data between subsequent K values was estimated according to Evanno et al, 200538 to identify the optimal number of clusters in the data. Outputs from STRUCTURE were graphically modified by DISTRUCT.39
Using results obtained from distance-based and model-based clustering method, significantly differentiated populations were chosen to represent source populations in the admixture calculation. ADMIX 2.040 was used to evaluate the estimator of admixture coefficient (mY) based on average coalescence times between pairs of genes sampled within and between populations.33 This method can use either the molecular distance between alleles or assume that all alleles are equally divergent. By using allele frequencies, the estimated coefficients are less affected by the stochasticity of the mutation process and the short time-scale of human admixture process is influenced by genetic drift rather than accumulated mutation.18, 41 Therefore, in this study, no divergence of alleles and no mutation model were used to estimate the genetic contribution of parental populations to the KM
Results
Genetic diversity
Although a detailed analysis of allele frequencies was not the objective of this study, some interesting points were observed (Table 2). The average observed heterozygosity (Ho) range (0.7360 (KM1)–0.8173 (KM4)) was similar to that previously reported for many Thai populations.42 This range was not statistically different among all the populations (χ2=12.5206; P=0.8622; Kruskal–Wallis test). The highest average heterozygosity of KM4 may have been shaped by the gene flow process, while the minimum average heterozygosity, found in KM1, might be the result of genetic drift. To confirm the relative effect of gene flow and genetic drift, a regression plot between average heterozygosity and distance from the centroid based on the model of Harpending and Ward (1982) reveals that the KM4 are the most displaced from the theoretical linear regression line, suggesting they have experienced the greatest amount of gene flow. KM1 were plotted lowest below from the line reflecting genetic drift, hence, less heterozygosity43 (Supplementary Figure 1). Three loci; D18S51 in KM1, D16S539 in LU1 and TH01 in SH; deviated from Hardy–Weinberg equilibrium, after applying Bonferroni correction. Allele frequency distributions, including several parameters such as power of discrimination, matching probability, polymorphic information content, power of exclusion and typical paternity index, are provided in supplementary materials (Supplementary Table 1–20).
Genetic affinity and genetic structure
To visualize the relationships among the populations, the MDS plot, derived from a distance matrix of Fst, is shown in Figure 2. The KM populations, except for KM7, are clustered together in the center of the MDS, with most of the Tai-speaking populations, indicating a close genetic relationship among Tai-speaking populations. The most likely explanation for the KM (except KM7) being located intermediately among the Tai-speaking populations is that the KM are admixed populations. For the outlier populations, LU2 and KM7 are the only 2, which separately plotted, from the 17 Tai populations, while the 3 Mon-Khmer populations (MO, LW1 and LW2) are segregated away from the center cluster as well as from each other, indicating high genetic differentiation. As found in the model-based clustering result, presented later in the text, the LW1, LW2 and LU2 initially emerge when the lower values of K are considered, confirming the great extent of genetic differentiation from any other populations (Figure 3).

Multidimensional scaling plot of studied populations based on Fst (filled circles: Tai-speaking populations, blank circles: Mon-Khmer-speaking populations).

Bar plot estimation figures of 10 putative parental populations, with sequential K from 2 to 7 (a–f) and their r values, inferred from the STRUCTURE 2.3 analyses. These plots were produced by the average of five replications, using DISTRUCT.
The model of isolation by distance predicts an increase in genetic differentiation with geographical distance, thus, correlation between genetic and geographical distances is expected.44 A Mantel test reveals an absence of correlation between genetic and geographical distances (r=0.2727, P=0.077), suggesting that factors in addition to geography describe the major trends of genetic variations among our populations.
Genetic differentiation of the parental populations
The 10 neighboring populations, which belonged to 7 ethnic groups, were considered as parental populations in admixture analysis, that is, indigenous Lawa (LW1 and LW2), the Mon (MO) and Tai-speaking populations (YU1, YU2, LU1, LU2, KH, YO and SH) which are ethnically, culturally and linguistically diversified through their historical migration and settlement. As the level of divergence between the parental populations is a crucial factor in admixture analysis,33 therefore, the clustering analysis was performed to choose the highly diverged populations among these 10 populations.
Based on pair-wise Fst, after adjusting the significance level according to the sequential Bonferroni method (Supplementary Table 21), KH-LU1, YU1-LU1, YU1-YO, YU1-SH and YO-SH were not significantly different. As STRUCTURE was unable to correctly identify the number of subpopulations at low levels of population differentiation (Fst <0.02),45 this limitation could be partially overcome by using the sampling location parameter of STRUCTURE.37 Because of the low level of genetic divergence (Fst ranges from 0.0028 to 0.0285) among our putative parental populations, the cluster analysis with the assistance of sampling information, which is more informative than those without (rMAX=0.39 in Figure 3), was chosen, and hence, only results from this model for K=1–7 are shown in Figure 3. By utilizing the ad hoc statistic ΔK, based on the rate of change in the log probability of data between successive K values,38 a strong modal peak at K=6 was found (data not show). Both populations with multiple sources of ancestry as well as populations with ancestry predominantly derived from one of the inferred components, are presented. In each K, individuals from the same population with similar ancestry showed high within-population genetic homogeneity.
At K=6, three distinct clusters of the Mon-Khmer-speaking groups were observed, although the MO cluster was not clearly delineated. The green and blue components in Figure 3 belonged to the highly diverged LW1 and LW2, respectively, while the orange component separated the MO from other populations. However, the MO cluster appears equally mixed by the yellow and red components from the Tai. Among the Tai-speaking populations, three main components, the yellow, purple and red, prevailed (Figure 3). The yellow component was commonly observed in KH and LU2, whereas purple characterized the LU1 population although LU1 was equally mixed with yellow. Finally, the red component distinguished the YU1, YO and SH from other populations. The YU2 seems to be more admixed with various Mon-Khmer and Tai components.
Admixture estimation
Based on genetic distance and STRUCTURE results, six highly differentiated populations were selected and divided into two linguistic groups; the Mon-Khmer (LW1, LW2 and MO) and the Tai (LU1, LU2 and YO), to represent variation present in parental populations.
The admixture proportions in different KM populations (Table 3) revealed that, when examining the genetic contribution from two parental populations, the Mon-Khmer contributed a lower proportion to almost all KM than did the Tai (Table 3 and Figure 4). Among the Mon-Khmer groups, the MO and LW2 contributed more genes to most of the KM than the LW1, which agrees with other historical evidence.2 Focusing on the contribution of the parental Tai, the YO arise as the major genetic contributor to most of the KM except KM3, KM5 and KM7, which have been more influenced by the LU. Variation in the KM genetic admixture patterns was observed. The study of admixture patterns suggested that gene flow from Tai-speaking populations to the KM was influenced more by the mass migration around 1800 AD, than by the migration in 1300 AD.

Pie diagrams showing the distribution of Mon-Khmer-speaking population (black) and Tai-speaking population (white) contributing to the Khon Mueang samples.
Discussion
This study aimed to analyze the population affinity among the present-day KM and their neighboring populations, and the genetic admixture of KM along the historical Yuan migration route by using a battery of 15 highly informative bi-parental STR loci. We hypothesize that the admixed origin of the KM was from the neighboring Mon-Khmer and Tai populations. In general, the admixed populations show genetic characteristics that are intermediate between the parental populations, although some admixed populations did not show this characteristic because of the genetic drift, as it was observed in previous reports.33, 46 The genetic diversity of all KM (Table 2) were within the same range as those of the neighboring populations, except for the average heterozygosity values of KM4 and KM1 which can be explained by the effect of gene flow and genetic drift, respectively. The MDS plot derived from allele frequency variance revealed that the KM populations, except for KM7, were located intermediately among the Tai-speaking populations (Figure 2), supporting their admixed origin.
The MDS plot also showed that the KM are more closely related to the neighboring Tai than the Mon-Khmer. The clustering as shown in the center of the plot between KM and their neighboring Tai indicated the linguistic population grouping in the Tai, but not in the Mon-Khmer-speaking groups. No clear geographical population grouping in the studied populations, and the absence of a correlation between genetics and geography was also observed. This implies that geographical factors have had less influence in shaping genetic variation in these populations.
The model-based clustering method, implemented in STRUCTURE, has been recognized as a powerful tool in characterizing population differentiation.45 We applied it here to estimate the extent of genetic differentiation among the parental populations. Generally speaking, samples from the Mon-Khmer groups (MO, LW1 and LW2), were remarkably genetically differentiated from each other as well as from the Tai-speaking populations. A possible explanation relies on their history and geographical location. The MO are more admixed than the LW, as shown by the presence of the Tai components (yellow and red in Figure 3). The MO geographical location, which was closer to the Tai than the LW might be served as a reason why the MO gene pool was highly admixed (Figure 1). The western Highland LW (LW1) have been living in thickly forested mountains in Mae Hong Son province and are characterized by limited mobility and the strong preservation of their LW identity.47 Hence, inbreeding might have occurred, as shown by their low genetic variation, reflected by total allele and diversity indices values (Table 2). The eastern Lowland LW (LW2) seem to have had some contacts with the YU, as shown by the presence of some components (yellow and red in Figure 3) typical to the YU, as they used to live in the city of Chiang Mai before they migrated to the Bo Luang plateau in Hod, Chiang Mai province. Because of linguistic and geographical barriers, this might possibly lead to restricted genetic exchange among both of the LW groups.
Among the seven Tai-speaking populations, there were three primary clusters identified by STRUCTURE. Surprisingly, despite the geographical proximity of the LU1 and LU2, they appear to form two weakly differentiated clusters. Both LU populations also have shared genetic components (yellow in Figure 3), but LU2 seems to have maintained a degree of genetic identity by social isolation, which can be seen from their location in the MDS plot and is mirrored in the low number of total alleles and a low diversity indices value (Table 2 and Figure 2), whereas LU1 might have mixed with other Tai during their migration through Laos.7
The study of admixture patterns may reflect our understanding of historical and anthropological aspects of demographic migrations.20 As already mentioned, there are three main geographical regions along the YU migratory route. The Chiang Rai basin was inhabited by the native LW and it was later occupied by the YU.4 Although the MO have never been reported as having lived in this area, the KM in Chiang Rai (KM1) seem to show a detectable genetic contribution from the MO. The recent backward migration of KM1 from Chiang Mai to Chiang Rai might explain this phenomenon.
The demographic scenario in the Chiang Mai-Lamphun basin is more complex. Before the YU movement, the upper and lower parts of this basin were previously dominated by the LW and MO respectively, in different periods.4 Thus, the admixture results were in agreement with ethno-historical sources. The KM, residing in the upper and lower parts of the Chiang Mai-Lamphun basin, had rather high admixture proportions from the LW and MO, respectively. In the plain of the Mae Cham basin, where the KM (KM7) migrated from Chiang Mai 200 years ago, the KM7 displayed a contribution from all parental groups similar to other KM samples. However, as shown in the MDS plot (Figure 2), KM7 has greater genetic divergence relative to other KM samples. The distinct gene pool of the Mae Cham population might be explained by admixture between them and other surrounding populations. As there are many populations, particularly the hill-tribes, living in the Mae Cham basin, admixture estimation may not be reliable. The hill-tribes might be parental sources for KM7, which would make them highly differentiated, but unfortunately there is no such genetic data set available to analyze.
Different admixture patterns were observed in two KM villages from Lampang. KM9 inevitably shows the highest fraction of MO admixture, who ruled the region between 750 and 1300 AD,3, 4 whereas the KM10 show the highest contribution from the native LW. The KM10, who migrated from the city of Chiang Saen, settled in the present Chiang Rai province, within the last 200 years.48 They could have mixed with the LW in Chiang Rai before their migration and then later mixed with the MO in Lampang.
The admixture observations in this study are largely consistent with historical records from northern Thailand. The variability in the KM genetic admixture patterns even among villages located in the same geographical region indicates a high heterogeneity among KM populations (Figure 5). This might reflect variation in the timing of village formation, and the parental populations of the region, when admixture started, had a greater influence on shaping the current gene pool of the KM, than geographical factors. An earlier genetic study suggested that the KM migrated from southern or south-eastern China.7 Several populations lived in proximity in an area of southern China, leading to extensive genetic exchange among them.19, 20, 21, 49, 50, 51 Although we were unable to look for admixture and affinities with the modern descendants of the KM ancestral populations from outside Thailand, this study did reveal genetic admixture among the KM in northern Thailand. Moreover, the present results from our admixture analysis are consistent with the suggestion from previous studies by the same group,7 using a battery of 15 highly informative bi-parental STR loci. Further study using genetic markers with different modes of inheritance are needed to gain a deeper understanding of genetic admixture and the population history of the KM.

Pie diagrams showing the distribution of MO (black), LW1 (checkered), LW2 (white), LU1 (vertical stripe), LU2 (horizontal stripe) and YO (dot) contributing to the Khon Mueang samples.
References
Schliesinger, J. Ethnic Groups of Thailand Non-Tai-Speaking Peoples, (White Lotus Press: Bangkok, 2000).
Schliesinger, J Tai group of Thailand, Volume 1: Introduction and overview, (White Lotus Press: Bangkok, 2001).
Penth, H A brief history of Lanna: civilizations of north Thailand, (Silkworm Books: Chiang Mai, 2000).
Ongsakul, S History of Lan Na, (Silkworm Books: Chiang Mai, 2005).
Condominas, G From Lawa to Mon, from Saa' to Thai, (The Australian national University: Canberra, 1990).
Gordon, R G Ethnologue: Languages of the World, 15th edn (SIL International: Dallas, 2005).
Kampuansai, J., Bertorelle, G, Castri, L., Nakbunlung, S., Seielstad, M. & Kangwanpong, D. Mitochondrial DNA variation of Tai speaking peoples in Northern Thailand. Sci. Asia 33, 443–448 (2007).
Bhumisak, J. The origin of Siam, Tai, Laos and Khmer (in Thai) (Textbook project foundation: Bangkok, 1990).
Charoenmuang, T. Khon Mueang, (Local government studied project)Faculty of Social Sciences, Chiang Mai University: Chiang Mai, 2001.
Nakbunlung, S. Origins and biological affinities of the modern Thai population: an osteological perspective, (Ph.D Thesis, University of Illinois: Urbana, 1994).
Poolsuwan, S. Science and the reality in Thai culture (in Thai) (Kob Fi: Bangkok, 2001).
Parra, E. J., Kittles, R. A., Argyropoulos, G., Pfaff, C. L., Hiester, K., Bonilla, C. et al. Ancestral proportions and admixture dynamics in geographically defined African Americans living in South Carolina. Am. J. Phys. Anthropol. 114, 18–29 (2001).
Parra, E. J., Marcini, A., Akey, J., Martinson, J., Batzer, M. A., Cooper, R. et al. Estimating African American admixture proportions by use of population-specific alleles. Am. J. Hum. Genet. 63, 1839–1851 (1998).
Bortolini, M. C., Da Silva, W. A., Junior, W., De Guerra, D. C., Remonatto, G., Mirandola, R. et al. African-derived South American populations: a history of symmetrical and asymmetrical matings according to sex revealed by bi- and uni-parental genetic markers. Am. J. Human. Biol. 11, 551–563 (1999).
Sans, M., Weimer, T. A., Franco, M. H., Salzano, F. M., Bentancor, N. & Alvarez, I. Unequal contributions of male and female gene pools from parental populations in the African descendants of the city of Melo, Uruguay. Am. J. Phys. Anthropol. 118, 33–44 (2002).
Sans, M. Admixture studies in Latin America: From the 20th to the 21st century. Hum. Biol. 72, 155–177 (2000).
Dupanloup, I., Bertorelle, G., Chikhi, L. & Barbujani, G. Estimating the impact of prehistoric admixture on the genome of Europeans. Mol. Biol. Evol. 21, 1361–1372 (2004).
Belle, E. M., Landry, P. A. & Barbujani, G. Origin and evolution of the Europeans's genome: evidence from multiple microsatellite loci. Proc. R. Soc. B. 273, 1595–1602 (2006).
Yao, Y. G., Lu, X. M., Luo, H. R., Li, W. H. & Zhang, Y. P. Gene admixture in the silk road of China: evidence from mtDNA and melanocortin 1 receptor polymorphism. Genes Genet. Syst. 75, 173–178 (2000).
Wen, B., Xie, X., Gao, S., Li, H., Shi, H., Song, X. et al. Analyses of genetic structure of Tibeto-Burman populations reveals sex-biased admixture in southern Tibeto-Burmans. Am. J. Hum. Genet. 74, 856–865 (2004).
Wen, B., Li, H., Lu, D., Song, X., Zhang, F., He, Y. et al. Genetic evidence supports demic diffusion of Han culture. Nature 431, 302–305 (2004).
Zhivotovsky, L. A., Underhill, P. A., Cinnio∂lu, C., Kayser, M., Morar, B., Kivisild, T. et al. The effective mutation rate at Y chromosome short tandem repeats, with application to human population-divergence time. Am. J. Hum. Genet. 7, 50–61 (2004).
Li, S. L., Yamamoto, T., Yoshimoto, T., Uchihi, R., Mizutani, M., Kurimoto, Y. et al. Phylogenetic relationship of the populations within and around Japan using 105 short tandem repeat polymorphic loci. Hum. Genet. 118, 695–707 (2006).
Deka, R., Shriver, M. D., Yu, L. M., Heidreich, E. M., Jin, L., Zhong, Y. et al. Genetic variation at 23 microsatellite loci in 16 human populations. J. Genet. 78, 99–121 (1999).
Reddy, B. M., Naidu, V. M., Madhavi, V. K., Thangaraj, K., Langstieh, B. T., Venkataramana, P. et al. STR data for the AmpF/STR Profiler Plus loci among 27 population of different social hierarchy from southern part of Andhra Pradesh, India. Forensic Sci. Int. 149, 81–97 (2005).
Pandu, G., Gandhi, K. P., Sharma, J. D., Chaubey, G. & Thangaraj, K. Genetic profile of nine STR loci among Goud and Padmashali populations of Andhra Pradesh, India. Forensic Sci. Int. 157, 201–205 (2006).
Thangaraj, K., Chaubey, G., Singh, V. K., Reddy, A. G., Pavate, P. P. & Singh, L. Genetic profile of nine autosomal STR loci among Halakki and Kunabhi populations of Karnataka, India. J. Forensic Sci. 51, 190–192 (2006a).
Sielstad, M., Bekele, E., Ibrahim, M., Toure, A. & Traore, M. A view of modern human origins from Y chromosome microsatellite variation. Genome Res. 9, 558–567 (1999).
Excoffier, L., Laval, G. & Schneider, S. Arlequin ver. 3.0: an integrated software package for population genetic data analysis. Evol. Bioinform. Online 1, 47–50 (2005).
Zar, J. H. Biostatistical Analysis, 4th edn (Prentice Hall: New Jersey, 1999).
Harpending, H. C. & Ward, R. In: Biochemical Aspects of Evolutionary Biology, (eds Nitecki M.) 213–256, (University of Chicago Press: Chicago, 1982).
Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220 1967.
Bertorelle, G. & Excoffier, L. Inferring admixture proportions from molecular data. Mol. Biol. Evol. 15, 1298–1311 (1998).
Rice, W. R. Analyzing tables of statistical tests. Evolution 43, 223–225 (1989).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies.. Genetics 164, 1567–1587 (2003).
Hubisz, M., Falush, D., Stephens, M. & Pritchard, J. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resources 9, 1322–1332 (2009).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study.. Mol. Ecol. 14, 2611–2620 (2005).
Rosenberg, N. A. DISTRUCT: a program for the graphical display of population structure.. Mol. Ecol. Notes. 4, 137–138 (2003).
Dupanloup, I. & Bertorelle, G. Inferring admixture proportions from molecular data: Extension to any number of parental populations.. Mol. Biol. Evol. 18, 672–675 (2001).
Barbujani, G. DNA variation and language affinities.. Am. J. Hum. Genet. 61, 1011–1014 (1997).
Listman, J. B., Malison, R. T., Sughondhabirom, A., Yang, B. Z., Raaum, R. L., Thavichachart, N. et al. Demographic changes and marker properties affect detection of human population differentiation.. BMC Genet. 8, 21 (2007).
Krithika, S., Maji, S. & Vasulu, T. S. Geographic Contiguity, Patterns of Gene Flow and Genetic Affinity among the Tribes of Arunachal Pradesh, India. Int J Hum Genet. 7, 267–276 (2007).
Cavalli-Sforza, L. L., Menozzi, P. & Piazza, A. The history and geography of human genes, (Princeton University Press: Princeton, 1994).
Latch, E. K., Dharmarajan, G., Glaubitz, J. C. & Rhodes Jr, O. E. Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation.. Conserv. Genet. 7, 295–302 (2006).
Di Benedetto, G., Ergüven, A., Stenico, M., Castri, L., Bertorelle, G., Togan, I. et al. DNA Diversity and Population Admixture in Anatolia.. Am J Phys Anthropol 115, 144–156 (2001).
Nahhas, R. W. Sociolinguistic survey of Lawa in Thailand, (Survey Unit Department of Linguistics Faculty of Humanities Payap University: Chiang Mai, 2007).
Princess Maha Chaki Siridhorn anthropology center, 2005, [http://www.sac.or.th/ethnic].
Li, H., Cai, X., Winograd-Cort, E. R., Wen, B., Cheng, X., Qin, Z. et al. Mitochondrial DNA diversity and population differentiation in southern East Asia. Am J Phys Anthropol. 134, 481–488 (2007).
Su, B., Xiao, C. & Jin, L. Genetic history of ethnic populations in southwestern China. Recent Advances in Human Biology. 8, 57–67 (2001).
Li, H., Wen, B., Chen, S. J., Su, B., Pramoonjago, P., Liu, Y. et al. Paternal genetic affinity between western Austronesians and Daic populations. BMC Evol Biol. 8, 146 (2008).
Acknowledgements
We thank all blood donors and village chiefs for their participation. We are also grateful to the Population Genetics and Genetic Epidemiology, the Genome Institute of Singapore, Singapore for genotyping support, as well as for training in statistical analyses from Dipartimento di Biologia ed Evoluzione, Universita di Ferrara, Ferrara, Italy. This study was supported by Royal Golden Jubilee Ph.D. program (Grant No. PHD/0243/2547).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Kutanan, W., Kampuansai, J., Colonna, V. et al. Genetic affinity and admixture of northern Thai people along their migration route in northern Thailand: evidence from autosomal STR loci. J Hum Genet 56, 130–137 (2011). https://doi.org/10.1038/jhg.2010.135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.135
Keywords
This article is cited by
-
Forensic and genetic characterizations of diverse southern Thai populations based on 15 autosomal STRs
Scientific Reports (2022)
-
Close genetic relationship between central Thai and Mon people in Thailand revealed by autosomal microsatellites
International Journal of Legal Medicine (2021)
-
Genetic and linguistic correlation of the Kra–Dai-speaking groups in Thailand
Journal of Human Genetics (2015)
-
Forensic STR loci reveal common genetic ancestry of the Thai-Malay Muslims and Thai Buddhists in the deep Southern region of Thailand
Journal of Human Genetics (2014)
-
Correlation between the linguistic affinity and genetic diversity of Chinese ethnic groups
Journal of Human Genetics (2013)
