
Abstract
Torg–Winchester syndrome (OMIM 259600) is an autosomal recessive multicentric osteolysis disorder. Mutations in the gene for matrix metalloproteinase 2 (MMP2) are involved in its pathogenesis. This is the first report of Torg–Winchester syndrome in east Asians. A 31-year-old female Korean patient had the typical clinical phenotypes of the syndrome, including shortening of trunk and limbs and severe osteolysis resulting in extremely small hands and feet. In addition, she had cord compression at the cervico-medullary junction, as well as lumbar dural ectasia. Molecular analysis revealed a novel homozygous missense mutation of MMP2, c.1217G>A (p.G406D). Gelatin zymography demonstrated a complete loss of the MMP2 activity of the mutation. Our results provide insights into the clinical and radiological features and pathogenic mechanisms of the syndrome.
Similar content being viewed by others


Introduction
Torg–Winchester syndrome (OMIM 259600) is an autosomal recessive multicentric osteolysis predominantly involving hands and feet. The syndrome was originally defined as three separate entities that showed marked clinical and radiological overlap: Torg syndrome, Winchester syndrome and nodulosis–arthropathy–osteolysis (NAO) syndrome. These syndromes had common clinical features and pathogenic mechanism.1 Hence, they have recently been re-classified as a single entity, Torg–Winchester syndrome, with NAO syndrome as a variant. The clinical spectrum of the disorder includes multiple peripheral osteolysis, subcutaneous fibro-fatty nodules, arthropathy and progressive joint contracture, along with various associated features, such as short stature, coarse face, corneal opacities, gum hypertrophy, hyperpigmentation and hypertrichosis.2, 3, 4, 5, 6 In addition, a few spinal manifestations as a part of generalized osteoporosis of vertebrae with concurrent compression fractures were reported,6, 7 but no further critical evaluation has been performed.
The gene encoding zinc-dependent matrix metalloproteinase 2 (MMP2) has been identified as a causative gene for Torg–Winchester syndrome.8 So far, seven MMP2 mutations have been reported (Table 1). Two homozygous mutations, c.302G>A and c.1021C>A, were first identified in Saudi-Arabian families.8 Subsequently, two homozygous mutations, c.1210G>A and c.1488_1490delTGG, were found in Italian and Algerian families, respectively.9, 10 A compound heterozygous mutation, (c.302G>A)+(c.1357delC), was observed in an American patient.2 Most recently, two homozygous mutations, c.1732delA and c.658+2T>C, were identified in Turkish families.5, 11
In this study, we report a Korean patient with Torg–Winchester syndrome focusing on radiographic findings of its spinal abnormalities, as well as identification of a novel loss of function mutation in MMP2.
Materials and methods
The patient is a 31-year-old Korean female, the youngest of eight siblings, with healthy non-consanguineous parents. Only one sister had similar clinical symptoms, but died at the age of 17 years. The patient had grown uneventfully until 3 years of age, when she began to develop painful contracture of fingers and hyperextension of great toes. She had several episodes of leg fractures with progressive leg weakness. She stopped growing at 12 years of age, with a final height of 124.5 cm. She gave birth to a healthy baby.
For mutation analysis, DNA fragments of the MMP2 gene, corresponding to each exon and its flanking introns, were amplified by the PCR using newly designed primers (Supplementary Table 1). The amplified PCR products were purified and sequenced using an automatic sequencer ABI 3730xl DNA analyzer (Applied Biosystems, Carlsbad, CA, USA). Sequence data were compared with a reference sequence (GenBank: NG_008989.1).
The activity of the mutant MMP2 protein was assessed using sodium-dodecyl-sulphate-polyacrylamide gel electrophoresis gelatin zymography. Serum samples (5 μl) from the patient and an unaffected control were electrophoresed in the presence of 0.1% SDS in an 8% polyacrylamide gel containing 1 mg ml–1 gelatin. Subsequent steps were carried out as described previously.12
Results
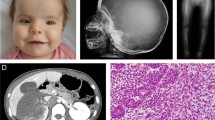
The clinical, radiological and genotypic features of our patient are summarized and compared with previously reported cases (Table 1). The patient showed extreme shortening of the trunk and limbs, fixed flexion of the elbows and wrists, stiff knees and hip adduction. All fingers and toes were foreshortened with telescoping of the soft tissue. A skeletal survey revealed generalized osteoporosis and osteolysis with destruction and resorption of the phalangeal, carpal and tarsal bones, as well as ankylosis of the elbow, hip and knee joints. She had osteoporosis with compression fractures at the lumbar spine (Figure 1a). She complained of progressive back pain and tingling sensations in the upper extremities. Magnetic resonance imaging showed osteolysis at the odontoid process of the axis with atlantoaxial subluxation, resulting in spinal cord compression (Figure 1b). Apparent widening of the spinal canal with dural ectasia and posterior scalloping of the lumbar vertebral bodies were also noted (Figure 1c).

Lateral radiograph and magnetic resonance imaging (MRI) of the spine of the patient. (a) Generalized severe osteoporosis of vertebrae with compression fracture of L4-5 vertebral bodies is noted. (b) Sagittal T2-weighted (T2W) MRI of the cervical spine shows osteolysis of the odontoid process, resulting in atlantoaxial subluxation and compression of the spinal cord at the cervico-medullary junction (arrow). (c) T2W image of the lumbar spine shows widening of the spinal canal with dural ectasia that brings posterior scalloping of the lumbar vertebral bodies (arrow).
We examined 13 exons of the MMP2 gene and identified a novel homozygous missense mutation, c.1217G>A (p.G406D), in exon 8 (Supplementary Figure 1). This mutation occurred in the α-helix B on the catalytic Zn2+-binding subdomain of the MMP2 protein,13 which is highly conserved across diverse species and in other members of the MMP protein family (Supplementary Figure 1). To demonstrate pathogenicity of this mutation, we assessed the serum MMP2 activity of the patient by a gelatin zymography. The results indicated complete loss of the gelatinolytic activity (Supplementary Figure 2).
Discussion
Spinal manifestations in Torg–Winchester syndrome have attracted little attention. Magnetic resonance imaging showed apparent osteolysis of the odontoid process, which resulted in atlantoaxial subluxation and compression of the cord at the cervicomedullary junction. The knowledge may provide valuable information in the aspect of prognosis and future management of the syndrome. Magnetic resonance imaging also provided information of dural ectasia through the lumbar spine that may cause progressive back pain in this patient. Thus, in practical aspect, spine evaluation with magnetic resonance imaging should be included as a part of skeletal survey in Torg–Winchester syndrome.
We identified a novel MMP2 mutation, c.1217G>A (p.G406D). The replaced glycine residue is located in the catalytic Zn2+-binding subdomain of MMP213 and evolutionally conserved (Supplementary Figure 1). The substitution of glycine with aspartic acid, an acidic amino acid would seriously affect the catalytic activity of MMP2 because of either loss of enzyme binding ability to Zn2+ or lowered affinity between the enzyme and Zn2+.
The molecular basis for the phenotypic spectrum of Torg–Winchester syndrome is unknown. It is interesting, the clinical and radiological features of our case, including the absence of subcutaneous nodules and wide metacarpals, were more similar to those of Winchester syndrome patients with homozygous mutations in the catalytic domain (p.V400del and p.E404K), than to those of NAO and Torg syndromes with other mutations (Table 1). This suggests that locations of MMP2 mutations may be associated with phenotypic characteristics of the syndrome. However, it is unclear how MMP2 inactivation alone could cause the phenotypic differences in Winchester, Torg and nodulosis–arthropathy–osteolysis syndromes, because so far, all known MMP2 mutations, either in the catalytic or non-catalytic domains have the same complete loss of the gelatinolytic activity.2, 8
Among the 23 known human MMP family proteins, there are five MMPs primarily involved in collagen and gelatin cleavage.13 MMP2 (gelatinase A) and MMP9 (gelatinase B) that have a unique catalytic domain are grouped into gelatinase; and MMP1 (collagenase 1), MMP8 (collagenase 2) and MMP13 (collagenase 3) are grouped into collagenase. It was recently reported that mutations in either MMP9 or MMP13 are responsible for the autosomal recessive form of metaphyseal anadysplasia (OMIM 613073).14 MMP13 mutations also cause spondyloepimetaphyseal dysplasia, Missouri type (OMIM 602111).15 Despite distinct phenotypes, these three disorders are caused by MMP family gene mutations. This may support the hypothesis that insufficient hydrolysis of gelatin or collagen in these disorders has a key role in the pathologic impairment of bone turnover, including bone formation and resorption.
References
Superti-Furga, A. & Unger, S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am J Med Genet A 143, 1–18 (2007).
Zankl, A., Pachman, L., Poznanski, A., Bonafe, L., Wang, F., Shusterman, Y. et al. Torg syndrome is caused by inactivating mutations in MMP2 and is allelic to NAO and Winchester syndrome. J Bone Miner Res 22, 329–333 (2007).
Phadke, S. R., Ramirez, M., Difeo, A., Martignetti, J. A. & Girisha, K. M. Torg-Winchester syndrome: lack of efficacy of pamidronate therapy. Clin Dysmorphol 16, 95–100 (2007).
Eisenstein, D. M., Poznanski, A. K. & Pachman, L. M. Torg osteolysis syndrome. Am J Med Genet 80, 207–212 (1998).
Tuysuz, B., Mosig, R., Altun, G., Sancak, S. & Glucksman, M. J. & Martignetti J.A. A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects. Eur J Hum Genet 17, 565–572 (2009).
Al Aqeel, A., Al Sewairi, W., Edress, B., Gorlin, R. J., Desnick, R. J. & Martignetti, J. A. Inherited multicentric osteolysis with arthritis: a variant resembling Torg syndrome in a Saudi family. Am J Med Genet 93, 11–18 (2000).
Winter, R. M. Winchester's syndrome. J Med Genet 26, 772–775 (1989).
Martignetti, J. A., Aqeel, A. A., Sewairi, W. A., Boumah, C. E., Kambouris, M., Mayouf, S. A. et al. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat Genet 28, 261–265 (2001).
Zankl, A., Bonafe, L., Calcaterra, V., Di Rocco, M. & Superti-Furga, A. Winchester syndrome caused by a homozygous mutation affecting the active site of matrix metalloproteinase 2. Clin Genet 67, 261–266 (2005).
Rouzier, C., Vanatka, R., Bannwarth, S., Philip, N., Coussement, A., Paquis-Flucklinger, V. et al. A novel homozygous MMP2 mutation in a family with Winchester syndrome. Clin Genet 69, 271–276 (2006).
Gok, F., Crettol, L. M., Alanay, Y., Hacihamdioglu, B., Kocaoglu, M., Bonafe, L. et al. Clinical and radiographic findings in two brothers affected with a novel mutation in matrix metalloproteinase 2 gene. Eur J Pediatr 169, 363–367 (2010).
Badier-Commander, C., Verbeuren, T., Lebard, C., Michel, J. B. & Jacob, M. P. Increased TIMP/MMP ratio in varicose veins: a possible explanation for extracellular matrix accumulation. J Pathol 192, 105–112 (2000).
Maskos, K. & Bode, W. Structural basis of matrix metalloproteinases and tissue inhibitors of metalloproteinases. Mol Biotechnol 25, 241–266 (2003).
Lausch, E., Keppler, R., Hilbert, K., Cormier-Daire, V., Nikkel, S., Nishimura, G. et al. Mutations in MMP9 and MMP13 determine the mode of inheritance and the clinical spectrum of metaphyseal anadysplasia. Am J Hum Genet 85, 168–178 (2009).
Kennedy, A. M., Inada, M., Krane, S. M., Christie, P. T., Harding, B., Lopez-Otin, C. et al. MMP13 mutation causes spondyloepimetaphyseal dysplasia, Missouri type (SEMD(MO). J Clin Invest 115, 2832–2842 (2005).
Acknowledgements
This work was supported by grants of the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea (A050234 and A080588).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Jeong, SY., Kim, BY., Kim, H. et al. A novel homozygous MMP2 mutation in a patient with Torg–Winchester syndrome. J Hum Genet 55, 764–766 (2010). https://doi.org/10.1038/jhg.2010.102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.102
Keywords
This article is cited by
-
Clinical, radiographic and molecular characterization of two unrelated families with multicentric osteolysis, nodulosis, and arthropathy
BMC Musculoskeletal Disorders (2023)
-
Bisphosphonates in multicentric osteolysis, nodulosis and arthropathy (MONA) spectrum disorder – an alternative therapeutic approach
Scientific Reports (2016)
-
Congenital generalized hypertrichosis: the skin as a clue to complex malformation syndromes
Italian Journal of Pediatrics (2015)
-
The ever-expanding conundrum of primary osteoporosis: aetiopathogenesis, diagnosis, and treatment
Italian Journal of Pediatrics (2014)
-
Functional characterisation of a novel mutation affecting the catalytic domain of MMP2 in siblings with multicentric osteolysis, nodulosis and arthropathy
Journal of Human Genetics (2014)
