
Abstract
A recent genome-wide association study by the International Multiple Sclerosis Genetics Consortium (IMSGC) reported association of 17 single-nucleotide polymorphisms (SNPs) in 14 loci with multiple sclerosis (MS). Only two loci, HLA-DRA and IL2RA, reached genome-wide significance (P<5E−08). In our study, we determined whether we could replicate the results of the IMSGC and whether more SNPs are genome-wide significantly associated with MS. We assessed the association between the 17 IMSGC SNPs and MS in three cohorts (total number of subjects 3981, among these 1853 cases). We performed a meta-analysis of the results of our study, the original IMSGC results and the results of a recent replication study performed in the Australian population. Of the 17 IMSGC SNPs, five SNPs showed genome-wide significant association with MS: HLA-DRA (P=8E−124), IL7R (P=6E−09), IL2RA (P=1E−11), CD58 (P=4E−09) and CLEC16A (P=3E−12). Therefore, genome-wide significance has now been shown for SNPs in different non-HLA MS risk genes. Several of these risk genes, including CD58 and CLEC16A, are shared by different autoimmune diseases. Fine mapping studies will be needed to determine the functional contributions to distinct autoimmune phenotypes.
Similar content being viewed by others


Introduction
Multiple sclerosis (MS) is a complex disease resulting from genetic and environmental factors. The genetic influence on MS susceptibility is substantial, as evidenced by the 20-fold increase in risk for siblings of patients. Much of the high recurrence risk is explained by the MHC Class II region.1 A recent genome-wide association (GWA) study2 conducted by the International Multiple Sclerosis Genetics Consortium (IMSGC) reported the association of MS with 17 single-nucleotide polymorphisms (SNPs) located in 14 regions. Only two regions, HLA-DRA and IL2RA, achieved genome-wide significance (P<5E−08). For a third gene, IL7R, convincing functional support was obtained2, 3, 4 and genome-wide significance was established in a joint analysis of 11 019 cases and 13 616 controls.5 In a recent Australian replication study of the 17 IMSGC risk SNPs, besides IL2RA, CLEC16A, RPL5 and CD58 were found to be associated with susceptibility for MS, although not genome-wide significant.6
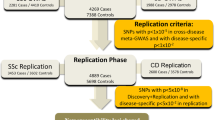
We here assessed MS association of the 17 IMSGC reported SNPs in MS patients from a Dutch genetically isolated population (45 cases and 195 controls), in MS patients from the Dutch general population (490 MS cases and 426 controls) and in MS patients from the Canadian Collaborative Project on the Genetic Susceptibility to MS (CCPGSMS; 1318 affected MS patients with their parents). In total, we studied 3981 subjects, including 1853 MS affected individuals. Results obtained in this study were also pooled with those obtained in the original IMSGC study2 and the recent Australian replication study.6
Materials and methods
Patients and genotyping
All patients fulfilled either Poser's criteria for definite MS or McDonald's criteria for MS.
The Dutch outbred cohort consisted of MS patients who are part of an ongoing nationwide study on genetic susceptibility in MS. A total of 490 MS patients were included, 370 sporadic MS patients and 120 cases from 120 multiplex MS families (that is, parents with two or more affected offspring). Overall, 10% of the patients (n=51) had a clinically isolated syndrome at the time of enrollment. The 426 healthy controls consisted of 26 unrelated spouses, together with 400 healthy blood donors. Further, we have sampled 45 MS patients within the framework of Genetic Research in Isolated Populations (GRIP) program.7 As controls, we included 195 healthy individuals from the same area who were all distantly related. Details on ascertainment are given elsewhere.8 A total of 1318 individuals with definite MS and 1507 of their unaffected first-degree relatives were typed as part of the CCPGSMS.9 The research protocol was approved by the respective Medical Ethics Committees and written informed consent had been obtained.
Genotyping was carried out using the MassARRAY system/Homogeneous MassExtend assay, following the protocol provided by Sequenom. PCR and extension primers were designed using the Assay Design 3.0 program (Sequenom, San Diego, CA, USA). Briefly, 20 ng genomic DNA is PCR amplified using Titanium Taq DNA Polymerase (Clontech, Mountain View, CA, USA). PCR primers were used at 100 nM final concentrations for a PCR volume of 10 μl. The PCR condition was 95 °C for 15 min, followed by denaturing at 94 °C for 20 s, annealing at 56 °C for 30 s, extension at 72 °C for 1 min for 45 cycles and finally incubation at 72 °C for 3 min. PCR products were first treated with shrimp alkaline phosphatase (Sequenom) for 20 min at 37 °C to remove excess dNTPs. ThermoSequenase (Sequenom) was used for the base extension reactions. Analysis and scoring were performed using the program Typer 3.3 (Sequenom).
Statistical analysis
All analyses were performed using R software (http://www.r-project.org/). Estimates of odds ratios (ORs) and significance was tested using logistic regression as implemented in ‘glm’ function. Analysis was performed without including covariates, therefore, effectively the analysis is equivalent to the Armitage trend test for proportions in genotypic 2 × 3 table. Thus, our analysis was similar to and compatible with those performed in external cohorts included in this meta-analysis, in that no adjustment was done for covariates and allelic ORs were estimated.
In the genetically isolated population, over-dispersion of the standard errors was estimated and corrected using genomic control approach.10 Genomic control lambda was estimated as 1.37.11 Meta-analysis of log (OR) was performed using a fixed model approach with inverse of the square of the estimates of standard error used as weights. Test for the heterogeneity of effects between studies was performed using standard Cochrane's Q-test; random effect model was estimated using ‘rmeta’ library for R (by T Lumley: http://cran.r-project.org/web/packages/rmeta/rmeta.pdf). We used a P-value of 5E−08 as the threshold for genome-wide significance.
The combined predictive value of the multiple genetic variants was investigated in a simulation study. The methods have been described elsewhere,12 but briefly, the simulation strategy creates a data set that includes genotypes and disease status for 100 000 individuals in such way that all genotype frequencies and ORs are the same as reported in this paper and the disease prevalence is 1 in 1000. Predicted risks for all individuals are obtained using Bayes’ theorem in which the earlier risk of disease (1 in 1000) is multiplied by the likelihood ratios of all single variants under the assumption of independent genetic effects. For this reason, we included one polymorphism per gene, selecting the polymorphism with the strongest OR. Therefore, in total we included 14 of the 17 SNPs. We examined the discriminative accuracy, which is the extent to which test results can discriminate between individuals who will develop MS and those who will not, and is commonly assessed by the area under the receiver operating characteristic curve (AUC). The AUC is the probability that the test correctly identifies the diseased individual from a pair, of whom one is affected and one is unaffected, and ranges from 0.5 (total lack of discrimination) to 1.0 (perfect discrimination). To obtain more robust AUC estimates, the simulation study was repeated 100 times and 95% confidence intervals were calculated. The AUC was obtained as the c-statistic by the function somers,2 which is available in the Hmisc library of R software (Harrell FE. Design and Hmisc R function library. Available at: http://biostat.mc.vanderbilt.edu/twiki/bin/view/Main/RS).
Results
Results of the meta-analysis are summarized in Table 1, which provides ORs and P-values for each study and for the combined analysis of all studies. The Supplementary Table 1 additionally provides allelic frequencies in cases and controls (where available) and random effect model meta-analysis.
The SNP rs3135388A located in the HLA-DRA region was most strongly associated with MS in all cohorts, as expected (Table 1). Meta-analysis, including the original IMSGC study, resulted in an OR estimate of 2.1 and a pooled P-value was 8E−124.
The SNP located in IL7R (rs6897932C) was consistently confirmed in the three populations and the meta-analysis reached genome-wide significance (P=6E−09). In addition, two SNPs within the IL2RA gene rs2104286T and rs12722489C reached genome-wide significance. Moreover, our analysis established two other SNPs with genome-wide significance. For rs12044852C located in CD58, the OR was 1.23 (P=4E−09) and for rs6498169G located in the CLEC16A locus (also named KIAA0350), the OR was 1.17 (P=3E−12). Forest plots for these SNPs are shown in Figures 1a and b. There is some support in our analysis that another locus rs10735781G in EVI5 is associated with MS (OR=1.12, P=2E−06), as independent replication was observed (Figure 1c) and the P-value of the meta-analysis improved compared with that reported in the initial IMSGC report. However, the overall analysis did not reach genome-wide significance (Table 1). Meta-analysis of SNPs in two genes that are in close proximity to EVI5, RPL5 and FAM69A, as well as SNPs in three other genes, ALK, CBLB and KLRB1, all did not change the P-values observed by the IMSGC substantially. For three loci (PDE4B, DBC1 and ANKRD15), our results suggest that the previously reported association in the initial IMSGC screen was a false positive association.

(a) Association between CD58 rs12044852[C] and MS. (b) Association between CLEC16A rs6498169[G] and MS. (c) Association between EV15 rs10735781[G] and MS. MS, multiple sclerosis. Effect estimates and 95% confidence intervals are shown. Study names: NEJM: IMSGC study; Dutch Outbred; GRIP: Dutch genetically isolated population; Oxford: Canadian population; Australian study.
Among genome-wide significantly implicated variants, two (rs6897932 at IL7R and rs3135388 at HLA-DRA) showed nominally significant heterogeneity (Table 1). The source of heterogeneity for rs6897932 may be the data of Australian study, which shows risk allele OR that is opposite (though not significant) to other studies. For the rs3135388, the heterogeneity may be introduced by the original study,2 which shows relatively low OR (1.99) and the Dutch Isolated population, showing a relatively high OR (3.08).
We calculated the AUC of the 14 genes (HLA included), found to be associated with MS by the IMSGC, for MS with the ORs we obtained in our meta-analysis. The AUC was 0.68 (95% confidence interval 0.67–0.68). The AUC for HLA alone was 0.63 (95% confidence interval 0.63–0.64), the AUC of the other 13 genes was 0.60 (95% confidence interval 0.59–0.61).
Discussion
These data further establish the genome-wide significant association with MS of 6 out of the 17 SNPs that were found to be associated with MS in a previous GWA study.2 The overall ORs of the non-HLA SNPs were modest, between 1.16 and 1.23. The predictive power of the 14 SNPs together was too low to have clinical significance, for example for diagnostic or prognostic purposes. The AUC is comparable with that found in recent studies on the prediction of type 2 diabetes based on 18 susceptibility genes and on the genetic prediction of coronary heart disease.13 The discriminative accuracy of HLA alone was better than the discriminative value of the other 13 genes together (0.63 vs 0.60).
Also, in the separate populations, ORs were generally below 1.30, with an exception of an 1.58 OR for the CD58 SNP in the Dutch outbred population and an OR of 1.96 for the replicated EVI5 SNP in the very small population of the genetic isolate.8 This study and others found that the risk effect from the HLA locus is independent of the risk signal coming from the other non-HLA risk loci.2, 6
We found genome-wide significant association of rs12044852 in the CD58 gene with MS (overall P=4E--09), which is in line with two very recent studies. The Australian and New Zealand Multiple Sclerosis Genetics Consortium showed genome-wide significance for the CD58 SNP rs1335532.14 This SNP is in strong linkage disequilibrium (LD) with rs12044852 (R2=0.93), both being located in intron 10/11 of the CD58 gene.15 In addition, a study in MS patients from the United Kingdom and the United States showed genome-wide significance, and further fine mapping indicated rs2300747 as the best susceptibility allele within the CD58 locus.16, 17 Todd and colleagues18 recently screened the rs12044852 SNP also in type 1 diabetes (T1D) patients in whom it does not seem to have a role. CD58 encodes a ligand for the T-cell specific CD2 membrane molecule, an adhesion molecule that transduces important signals for T-cell proliferation and differentiation. In addition, a role for the CD58 molecules has been suggested in chronic inflammatory polyneuropathies.19
The CLEC16A rs6498169 SNP (intron 22) was also found to be genome-wide significantly associated to MS when combining the Australian study6 and the IMSGC screen (P=3E−08).2 Genome-wide significance was further only noted for another SNP in this gene, the T1D-associated SNP rs12708716 located in intron 19, (P=1.6E−16).20 In the recently published meta-analysis and replication study in MS patients from the UK and the US, the SNP rs11865121 in the CLEC16A gene was found to be associated with susceptibility to MS (P=1.77E−07) in the joint analysis.17 A study in Sardinia that explored the contribution of T1D genes to MS risk, found association, although not genome-wide significant, with another SNP in intron 19 rs725613 (P=4E−05),21 which is in perfect LD with the genome-wide significantly associated SNP rs12708716 (R2=1.0). The CLEC16A SNP rs6498169 that was identified here as genome-wide significant is in a different haplotype block (R2=0.2), suggesting that in a single gene different SNPs are involved to different autoimmune disorders. A third autoimmune disorder that has been associated with CLEC16A is autoimmune Addison’s disease.22 It remains to be determined to what extent the different genetic variants within the CLEC16A area contribute to the susceptibility for certain autoimmune disorders.
Still little is known regarding the function of CLEC16A protein in humans. It is a member of the C-type lectin family, of which members have described to provide signals for a decision between tolerance and immunity. They can bind bacterial products as well as endogenous ligands, and their signal can counteract the signal of Toll-like receptors, therewith influencing T-helper cell function. We previously implied a role for C-type lectin receptors in MS pathogenesis, and discussed a link with infections.23 However, further research has to be undertaken to understand the exact function of the CLEC16A gene and subsequently how it could influence the susceptibility to MS.
Not surprisingly, many of the risk genes identified thus far are directly linked to adaptive immune functions, further stressing the autoimmune pathogenesis of the disease. IL2R and IL7R are receptors for the regulation of lymphocyte expansion and differentiation. CD58 and CLEC16A both share functional characteristics with the recently identified MS risk gene CD22620, 24, 25 that reached genome-wide significance,20 by their involvement in cell–cell interaction, adhesion and signaling.
So far KIF1B has been reported the only neuronally expressed gene with genome-wide significant association with MS.11
It is of note that many by now validated and strongly suggested MS susceptibility loci are also associated with other autoimmune diseases (AID) such as type I DM and Graves’ disease. Although these genes may very well account for the clustering of MS and other AID in certain populations and within families, here again the protective as well as risk alleles MHC class II area may exert the strongest effects.
Some immunomodulatory treatments can trigger AID such as Graves’ disease or idiopathic thrombocytopenic purpura in MS patients.26, 27 Genotyping of the overlapping autoimmune risk alleles24 may identify patients at risk for such autoimmune side effects.
In conclusion, in this study genome-wide significant association of non-HLA risk genes with susceptibility to MS is confirmed for CD58, CLEC16A, IL2R and IL7R. Several of the by now validated and strongly suggested risk genes are shared by different AID, including the risk genes for which we found genome-wide significance. A question that remains to be answered is whether the SNPs in these are causative variants. Fine mapping studies will be needed to determine the functional contributions to the distinct autoimmune phenotypes.
Conflict of interest
The authors declare no conflict of interest.
References
Lincoln, M. R., Montpetit, A., Cader, M. Z., Saarela, J., Dyment, D. A., Tiislar, M. et al. A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nat. Genet. 37, 1108–1112 (2005).
Hafler, D. A., Compston, A., Sawcer, S., Lander, E. S., Daly, M. J., De Jager, P. L. et al. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 357, 851–862 (2007).
Lundmark, F., Duvefelt, K., Iacobaeus, E., Kockum, I., Wallstrom, E., Khademi, M. et al. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat. Genet. 39, 1108–1113 (2007).
Gregory, S. G., Schmidt, S., Seth, P., Oksenberg, J. R., Hart, J., Prokop, A. et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet. 39, 1083–1091 (2007).
International Multiple Sclerosis Genetics Consortium (IMSGC). Refining genetic associations in multiple sclerosis. Lancet Neurol. 7, 567–569 (2008).
Rubio, J. P., Stankovich, J., Field, J., Tubridy, N., Marriott, M., Chapman, C. et al. Replication of KIAA0350, IL2RA, RPL5 and CD58 as multiple sclerosis susceptibility genes in Australians. Genes Immun. 9, 624–630 (2008).
Aulchenko, Y. S., Heutink, P., Mackay, I., Bertoli-Avella, A. M., Pullen, J., Vaessen, N. et al. Linkage disequilibrium in young genetically isolated Dutch population. Eur. J. Hum. Genet. 12, 527–534 (2004).
Hoppenbrouwers, I. A., Cortes, L. M., Aulchenko, Y. S., Sintnicolaas, K., Njajou, O., Snijders, P. J. et al. Familial clustering of multiple sclerosis in a Dutch genetic isolate. Mult. Scler. 13, 17–24 (2007).
Sadovnick, A. D., Risch, N. J. & Ebers, G. C. Canadian collaborative project on genetic susceptibility to MS, phase 2: rationale and method. Canadian Collaborative Study Group. Can. J. Neurol. Sci. 25, 216–221 (1998).
Bacanu, S. A., Devlin, B. & Roeder, K. The power of genomic control. Am. J. Hum. Genet. 66, 1933–1944 (2000).
Aulchenko, Y. S., Hoppenbrouwers, I. A., Ramagopalan, S. V., Broer, L., Jafari, N., Hillert, J. et al. Genetic variation in the KIF1B locus influences susceptibility to multiple sclerosis. Nat. Genet. 40, 1402–1403 (2008).
Janssens, A. C., Aulchenko, Y. S., Elefante, S., Borsboom, G. J., Steyerberg, E. W. & van Duijn, C. M. Predictive testing for complex diseases using multiple genes: fact or fiction? Genet. Med. 8, 395–400 (2006).
Janssens, A. C. & van Duijn, C. M. Genome-based prediction of common diseases: advances and prospects. Hum. Mol. Genet. 17 (R2), R166–R173 (2008).
Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene). Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosome 12 and 20. Nat. Genet. 41, 824–828 (2009).
Baranzini, S. E., Wang, J., Gibson, R. A., Galwey, N., Naegelin, Y., Barkhof, F. et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum. Mol. Genet. 18, 767–778 (2009).
de Jager, P. L., Baecher-Allan, C., Maier, L. M., Arthur, A. T., Ottoboni, L., Barcellos, L. et al. The role of the CD58 locus in multiple sclerosis. Proc. Natl Acad. Sci. USA 106, 5264–5269 (2009).
de Jager, P. L., Xiaoming, J., Wang, J., de Bakker, P. I. W., Ottoboni, L., Aggarwal, N. T. et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 41, 776–782 (2009).
Fung, E., Smyth, D. J., Howson, J. M., Cooper, J. D., Walker, N. M., Stevens, H. et al. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun. 10, 188–191 (2009).
van Rhijn, I., van den Berg, L. H., Bosboom, W. M., Otten, H. G. & Logtenberg, T. Expression of accessory molecules for T-cell activation in peripheral nerve of patients with CIDP and vasculitic neuropathy. Brain 123, 2020–2029 (2000).
International Multiple Sclerosis Genetics Consortium (IMSGC). The expanding genetic overlap between multiple sclerosis and type I diabetes. Genes Immun. 10, 11–14 (2009).
Zoledziewska, M., Costa, G., Pitzalis, M., Cocco, E., Melis, C., Moi, L. et al. Variation within the CLEC16A gene shows consistent disease association with both multiple sclerosis and type 1 diabetes in Sardinia. Genes Immun. 10, 15–17 (2009).
Skinningsrud, B., Husebye, E. S., Pearce, S. H., McDonald, D. O., Brandal, K., Wolff, A. B. et al. Polymorphisms in CLEC16A and CIITA at 16p13 are associated with primary adrenal insufficiency. J. Clin. Endocrinol. Metab. 93, 3310–3317 (2008).
‘t Hart, B. A., Laman, J. D., Bauer, J., Blezer, E., van Kooyk, Y. & Hintzen, R. Q. Modelling of multiple sclerosis: lessons learned in a non-human primate. Lancet Neurol. 3, 588–597 (2004).
Hafler, J. P., Maier, L. M., Cooper, J. D., Plagnol., V., Hinks, A., Simmonds, M. J. et al. CD226 Gly307Ser association with multiple autoimmune diseases. Genes Immun. 10, 5–10 (2009).
Seldin, M. F. & Amos, C. I. Shared susceptibility variations in autoimmune diseases: a brief perspective on common issues. Genes Immun. 10, 1–4 (2009).
Coles, A. J., Wing, M., Smith, S., Coraddu, F., Greer, S., Taylor, C. et al. Pulsed monoclonal antibody treatment and autoimmune thyroid disease in multiple sclerosis. Lancet 354, 1691–1695 (1999).
Samijn, J. P., te Boekhorst, P. A., Mondria, T., van Doorn, P. A., Flach, H. Z., van der Meche, F. G. et al. Intense T cell depletion followed by autologous bone marrow transplantation for severe multiple sclerosis. J. Neurol. Neurosurg. Psychiatr. 77, 46–50 (2006).
Acknowledgements
This study was supported by grants from MS Research Netherlands (RQH and CvD), the Netherlands Organisation for Scientific Research (ZON-MW, RQH), Erasmus MC and the Multiple Sclerosis Society of Canada Scientific Research Foundation and the Multiple Sclerosis Society of the United Kingdom (SVR, GCE). The GRIP study is supported by Centre for Medical Systems Biology (CMSB). We are grateful to all patients and their relatives, general practitioners and neurologists for their contributions and to P Veraart, genealogist in the GRIP area, for her help in genealogy. D Lont and the personnel of the VIB Genetic Service Facility (http://www.vibgeneticservicefacility.be/) for genotyping and P Snijders, general practitioner in the GRIP area, for his help in data collection. Our study complies with the current laws of the countries in which they were performed.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website (http://www.nature.com/jhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Hoppenbrouwers, I., Aulchenko, Y., Janssens, A. et al. Replication of CD58 and CLEC16A as genome-wide significant risk genes for multiple sclerosis. J Hum Genet 54, 676–680 (2009). https://doi.org/10.1038/jhg.2009.96
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.96
Keywords
This article is cited by
-
Association of CD58 polymorphism and multiple sclerosis in Malaysia: a pilot study
Autoimmunity Highlights (2019)
-
Serum of patients with acute myocardial infarction prevents inflammation in iPSC-cardiomyocytes
Scientific Reports (2019)
-
Do Human Endogenous Retroviruses Contribute to Multiple Sclerosis, and if So, How?
Molecular Neurobiology (2019)
-
CD58 polymorphisms associated with the risk of neuromyelitis optica in a Korean population
BMC Neurology (2014)
-
Genetic variants in IL2RA and IL7R affect multiple sclerosis disease risk and progression
neurogenetics (2014)
