
Abstract
Central areolar choroidal dystrophy (CACD) is a rare inherited disease, which causes progressive profound loss of vision in patients during their fourth decade. It is characterized by atrophy of retinal pigment epithelium, photoreceptors and choriocapillaris. The disease showed a genetic heterogeneity. Previously, mutations in the peripherin/RDS gene and a linkage to the CACD locus in the 17p13 region have been reported in CACD families. In this study, we report on a clinical and genetic investigation of CACD in a large Tunisian consanguineous family with 21 affected individuals in three living generations. CACD has been associated with drusen in some of them. Linkage analysis and mutational screening exclude linkage to the PRPH2/RDS gene and to the CACD locus. These data provide further evidence of the genetic heterogeneity of CACD.
Similar content being viewed by others

Introduction
Central areolar choroidal dystrophy (CACD; MIM%215500) is a progressive macular dystrophy characterized by subtle, mottled depigmentation in the posterior pole in the early stages.1 The depigmentation area gradually enlarges until an oval or round surface of atrophy of the retinal pigmentary epithelium and choriocapillaris is formed,2 as described by histopathological studies.3 Typically, no flecks or drusens are observed in this chorioretinal dystrophy.4 The disease starts between the third and fifth decade of life, with a fine, at times hardly detectable, mottling of the parafoveal pigment epithelium. Later, between the fourth and seventh decade of life, progressive macular atrophy leads to a dramatic decline in central visual acuity in most cases. Early lesions consist of a granularity next to the fovea and the pathognomonic areolar zone of atrophy develops over the following decades.5 CACD may show an autosomal recessive or dominant inheritance.6
Defined mutations in the PRPH2 gene (Argl72Trp, Argl72Gln, Argl42Trp and Arg195Leu) are known to cause progressive CACD,7, 8, 9, 10 although other, as yet unidentified, genetic causes may underlie this condition. Recently, a second locus at chromosome 17p13 has also been identified by a genome-wide linkage search in a large Northern Irish family with a type of autosomal dominant CACD.11 It maps to a 5 cM region within the D17S1810 and CHLC GATA7BO3 interval.12
In this study, we report on a large Tunisian CACD family consisting of 21 patients, of whom six presented with drusen, in which we carried out a genetic investigation. We excluded linkage to the PRPH2 and CACD loci, thus underlying the genetic heterogeneity of this chorioretinopathy.
Materials and methods
Clinical investigation
The proband of the CACD family (CACD8) presented to the Department of Ophthalmology in La Rabta Hospital for an ophthalmological examination because of a complaint of low vision in the seventh decade. The questionnaire including a pedigree drawing identified several cases with visual complaints among his relatives. For this purpose, we visited the large family at home and established a first diagnosis using ophthalmic portable equipment.
We identified and examined a three-generation family with CACD originating from North Eastern Tunisia (Figure 1). Diagnosis, undertaken in the hospital a second time, was based on a clinical ophthalmic examination including visual acuity measurement, slit lamp examination of the anterior segments, ocular pressure measurement by Goldmann applanation tonometry, vitreous examination, complete fundus examination, fluorescein angiography and Humphrey visual field examination.

Pedigree structure of the Tunisian central areolar choroidal dystrophy (CACD) family and its haplotype analysis at the PRPH2/RDS and CACD locus. For the sake of clarity and because of the large family size, we partially represent here the genealogy. A bar above an individual indicates an individual who was clinically examined by the author. An asterisk above an individual indicates the presence of drusen in fundus examination. Marker names are shown on the left, according to the genomic physical map spanning the CACD locus, which was previously used by Hughes et al.12 and Lichanska et al.13 Markers present 80, 100, 69.2, 92 and 56.3% of heterogeneity, respectively (top to bottom).14
Molecular genetic analysis
After informed consent was obtained, blood samples were taken for DNA extraction from peripheral blood leukocytes by standard phenol-chloroform procedures.15 PCR amplification reaction was carried out in a volume of 25 μl containing 10 ng of DNA, 10 × PCR buffer, 0.2 mM of each dNTP, 1.5 mM MgCl2, 0.5 μM of one of each primer and 0.5 U Amplitaq DNA polymerase (Invitrogen, Foster City, CA, USA). DNA was denatured for 5 min at 96 °C, followed by 40 cycles with denaturation at 94 °C for 30 s, annealing at the optimal annealing temperature depending on the primer pair used for 35 s, elongation at 72 °C for 40 s and terminal elongation for 10 min at 72 °C.
To test linkage to the PRPH2/RDS gene and to the 17p13 region, we typed all available members using two polymorphic markers, AFM136yf8 (D6S271) and AFM238zf4 (D6S426), and the three following polymorphic markers located on locus CACD spanning a 5 cM interval, AFMa282yd9 (D17S1810), AFM177xh6 (D17S796) and AFM070xg5 (D17S1353), which were selected from genetic maps in accordance with the constructed map of the CACD region.12, 13 Genotyping was carried out as described elsewhere.16
Mutation screening was performed for the three coding exons of the PRPH2 gene by direct sequencing using an ABI Prism 3130 genetic analyzer (Applied Biosystems, Foster City, CA, USA), in accordance with the manufacturer's recommendations. Primer sequences are available on request.
Statistical methods
The MERLIN v1.0.1 software program17 was used for computerized multipoint parametric analysis of data, assuming autosomal dominant inheritance with a disease allele frequency of 0.0001 and complete penetrance in members of generations I, II and III. Members of generation VI were assigned as affected or normal on the basis of the presence or absence of fundus abnormalities thought to be indicative of early disease to one of four age-related liability classes ranging from 80 to 95% likelihood of the assigned disease status. This was because diagnosis is not completely certain in young family members before the onset of pathognomonic features in the fourth decade.10
Results
Clinical data
We identified and examined a three-generation family with CACD (Figure 1 and Table 1). A total of 21 patients were found to be affected and six of them had a drusen (CACD5, CACD15, CACD16, CACD19, CACD21 and CACD24). Their findings, varying with disease state, agreed with the Hyong and Deutman classification18 (Table 1). The mean age at onset of visual loss was 47 years, with a subsequent gradual deterioration in visual acuity (Table 2). These are some instances of affected patients describing the evolution of CACD in this family (Figure 2).

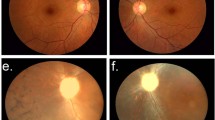
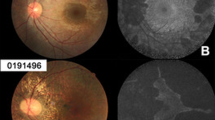
Fundus photographs and autofluorescence images showing central areolar choroidal dystrophy (CACD) disease progression throughout generations in the Tunisian family. (a and b) Stage I, Fluorescein angiography: parafoveaolar alterations of retinal pigmentary epithelium with punctiform hyperfluorescent lesions leading to a window effect. (c) Stage II, arteriovenous transit times: alternating radial hypofluorescent and hyperfluorescent lesions. (d) Stage II, late-phase angiography: depigmentation and atrophy of retinal pigment epithelium cells around the fovea, with pigmentary changes in temporal to the fovea. (e and f) Stage III, fundus examination and fluorescein angiography (before injection) in the same right eye. (g) Stage III, early-phase angiography. (h) Stage III, late-phase angiography. (i) Stage IV, fundus examination. (j) Stage IV, angiography arteriovenous transit times: retrograde filling of the affected vessel on the atrophic area. However, the big vessels are still visible. (k) Stage IV, angiography late phase, showing an impregnation in the choroid that occurred by soaking fluorescein mainly in the periphery than in the center of the lesion.
Case 1: CACD8 patient
This 70-year-old man is the index case. He presented to the Department of Ophthalmology for an evaluation of a decrease in visual acuity since the past 5 years. His visual acuity was 1/20 P20 in the right eye and 1/50 P20 in the left eye. An examination of the anterior segment revealed a bilateral corneal epithelial dystrophy and a bilateral cataract. His intraocular pressure (IOP) of 12 mm Hg was normal. Fundus examination revealed round and symmetric lesions of 3–4 gray papillary diameter in the macula underlying the atrophy and choroidal vessels. In each eye, there was a loss of foveal reflex, and the optic disk, retinal vessels and periphery were normal. Fluorescein angiography showed an atrophy of the pigment retinal epithelium and a loss of choriocapillaris. The hyperfluorescence of fatty choroidal vessels, which are clearly visible in the macula, was observed without diffusion of fluorescein throughout the lesion. In addition, we noticed a peripapillary hypofluorescence due to chorioretinal atrophy. His retinal periphery was normal. The visual field showed a bilateral central absolute scotoma. Two years later, he was reexamined and his visual acuity was then 1/50 P20 in the right eye. However, no evolution was noticed in the left eye and in the fundus examination. These clinical features correspond to the fourth and most advanced stage.
Case 2: CACD4 patient
This 49-year-old man is the CACD8 patient's son. His general health was excellent. He had a normal anterior segment. His best corrected visual acuity was 10/10 P2 in the right eye and 10/10 P2 in the left eye and his visual field was normal. He presented white and yellow lesions at the peripheral fovea in the fundus examination (Figure 2). His fluorescein angiography revealed a broadening of the central nonvascular area with a hyperfluorescence border that outlined the lesion with window defects corresponding to the first stage of CACD.
Case 3: CACD2 patient
This 51-year-old woman is the sister of the case reported previously. She had a normal anterior segment and her best visual acuity was 10/10 P2 in the right eye and 10/10 P2 in the left eye. Her visual field was unaffected. The fundus examination revealed a slight pigmentary reshuffle in both eyes, with some white dots under temporal foveolar lesions especially in the left eye. She had type II diabetes 5 years ago. No diabetic retinopathy was shown in fluorescein angiography. The foveolar region is hypofluorescent and becomes progressively hyperfluorescent away from the center. The diagnosis corresponds to the second stage of CACD.
Case 4: CACD13 patient
This 53-year-old man is the uncle of the two previous cases. While interviewing family members, he complained of a progressive decrease in visual acuity since the past 4 years. His visual acuity was 4/10 P5 in the right eye and 2/10 P5 in the left eye. A bilateral nuclear cataract was detected in the anterior segment. IOP was 14 mm Hg in both eyes. Fundus examination revealed round and symmetric lesions of 2–3 yellowish papillary diameter in the macula, highlighting atrophy and choroidal vessels. His foveolar reflex was normal.
Fluorescein angiography showed a well-demarcated loss of the retinal pigment epithelium (RPE) and a choriocapillaris atrophy. This area corresponds to a round-to-oval zone of absent fluorescence involving the fovea in late-stage CACD, bordered by a small residual band of increased fluorescence. The visual field revealed a bilateral central relative scotoma.
Two years later, this patient was reexamined in the Department of Ophthalmology. We noticed a moderate decrease of near vision using the Parinaud scale (P10) in both eyes. However, the far vision remained steady. Finally, the patient was diagnosed as affected at the third stage of CACD.
Haplotype and mutation analysis
On the basis of clinical features strongly suggesting the CACD phenotype and because of the relatively small size of the PRPH2 gene, a molecular investigation was initiated by screening individuals of two nuclear families (see Supplementary data).
Screening for mutations revealed Val106Val, Glu304Gln, Lys310Arg, Gly338Asp and de novo IVS3+15C>T variations, but none of the previously reported deleterious mutations or other mutations were observed in the PRPH2 gene. This result suggested that patients could be mutated in the unexplored regulatory region of the PRPH2 gene or in the excluded linkage to this gene. As the family was potentially informative for linkage analysis, all available family members were genotyped using microsatellite markers flanking the PRPH2 gene. Haplotype analysis showed that affected siblings were heterozygous for the two closest markers to the RDS gene, consistent with an autosomal dominant inheritance.
Affected individuals IV.1, IV.2, IV.12, IV.13 and IV.15 did not share a common disease haplotype. In addition, patient IV.3 had the same disease haplotype as her healthy sister IV.4. Furthermore, different haplotypes segregated with CACD in this family, thus giving evidence for an exclusion of the peripherin gene.
Failure to detect a linkage in the PRPH2 gene, a key candidate gene, prompted us to determine whether the CACD locus is involved in chorioretinopathy in this family.
For this purpose, a linkage analysis was performed. On the basis of a similar analysis approach, haplotyping showed that affected individual IV.3 and her healthy sister IV.4 shared the same haplotype and that different haplotypes segregated with CACD in this family. The haplotype analysis also revealed several recombinations between the tested markers and the CACD locus within this family. This is suggestive of an exclusion of linkage between the CACD locus and the morbid allele in this family, and underlies the genetic heterogeneity of the disease (Figure 1).
Multipoint logarithm of the odds ratio (LOD) scores were calculated for this family between the disease locus and the two loci (Table 3). Multipoint analysis clearly ruled out a linkage of the studied family to the PRPH2 gene and to the CACD locus (the value of the LOD score was <−2), thus confirming the results of the haplotype analysis and the exclusion of the involvement of these loci in CACD.
Discussion
Central areolar choroidal dystrophy is a clinically and genetically heterogeneous progressive macular dystrophy. Until now, this rare disease has been reported mainly in patients of European and Asian descent. This is, to our knowledge, the first study of CACD in Africa and in the Arab world. In this study, we report a clinical and genetic investigation of CACD in a three-generation Tunisian family. A total of 21 affected family members were examined at different stages of CACD disease progression. All had reduced central visual acuity, and a fundus and angiogram pattern consistent with the diagnosis of CACD disease. The differential diagnosis between cone dystrophy and CACD was carried out by the presence of large choroidal vessels characterizing CACD, which is rarely encountered in cone dystrophy.20
Although typically no flecks or drusens are observed in CACD,3 the clinical spectrum in the Tunisian family ranged from CACD without drusen in 15 individuals to the fully expressed phenotype of CACD with drusen in six patients. Among these, one patient had macular drusen (CACD16) in stage IV, and five (CACD5, CACD15, CACD19, CACD21 and CACD24) had drusen associated to RPE defects in stages I and II, thus illustrating a variable expression of CACD–drusen association. However, because drusen is recognized after the age of 20 years in crystalline retinal degeneration, Hutchinson-Tay choroiditis, Doyne's honeycomb choroiditis, Holthouse-Batten's superficial chorioretinitis and malattia leventinese, we could not ascertain whether drusen in our patients was also present at an early stage. Although CACD–drusen association was previously reported,18 it is still not fully elucidated. However, our series is older than Klevering's series and drusen is not always present in great numbers. It could be possible that drusen and CACD are variable expressions of the same disease, as a genetic study has demonstrated that these are allelic disorders to the PRPH2 gene.18 Taking into account the lack of choroidal neovessels in Tunisian patients and the relationship between the appearance of drusen and the evolution of CACD, drusen could likely be unrelated to CACD, which explained the development of drusen in some but not all CACD patients.
Although the follow-up time of two patients (CACD8 and CACD13) could be relatively short (2 years), CACD disease seems to have a slow course. However, legal blindness was found in four eyes. Strikingly, the disease course is likely to be more rapid in men than in women. This simultaneous occurence of a slow and rapid evolution has also been observed in the same family.21 This could be because of variable expressivity or influence of modifier genes under genomic imprinting.
In this family, CACD is inherited as an autosomal dominant trait across three generations. Taking into account the higher degree of consanguinity in this family, an autosomal recessive trait cannot be ruled out in the absence of clinical data of the patients’ ancestry. However, this latter hypothesis remains unlikely, as a healthy son (IV.9) was born in the nuclear family in whom both parents are affected (III.9 and III.10).
In the consanguineous family, we observed CACD added to cataract in three patients (CACD8, CACD13 and CACD23), added to type I diabetes in one patient (CACD2) and to type II diabetes in CACD5, CACD6, CACD14 and CACD21 patients, for whom fluorescein angiography showed diabetic retinopathy. The occurrence of such a clinical association was not previously described. We believe that these could be fortuitous, and likely age related.
Consanguinity could also concentrate autosomal recessive genes in the offspring, leading to additional disorders. This was observed in two non-CACD cases (IV.17 and IV.18); individual IV.18 had a severe motor impairment and mental retardation and his sister IV.17 presented a profound prelingual hearing loss and muteness. Although the perceptive hearing loss was previously described with an association to CACD,22 our observation in the studied family likely corresponds to a fortuitous association of separate clinical entities because of the high degree of consanguinity.
Age of onset, progression of disease and characteristic fundus abnormalities share similarities to previous reports on families with CACD associated with peripherin/RDS gene mutations. The Val106Val, Glu304Gln, Lys310Arg, Gly338Asp and IVS3+15C>T variations detected in Tunisian CACD patients are considered as nondeleterious variants and the assessed haplotype does not segregate with disease in this family. The exclusion of the CACD locus is also suggestive of an unknown responsible gene. Striking variability in individual phenotypic findings in our family suggests additional factors that modify the phenotypic expression of the disease.
Although CACD is a rare disorder, discovering an exclusion of the two known CACD-related locations in this family is important not only in relation to the disease but also as it may highlight genes that may be implicated in the pathology of age-related macular degeneration, which is currently classified among the priority eye diseases by the World Health Organization (WHO, World Human Organization http://www.who.int/blindness/causes/priority/fr/).
References
Nettleship, E. Central senile areolar choroidal atrophy. Trans. Ophthalmol. Soc. UK 4, 165–166 (1884).
Hyong, C. B. & Deutman, A. F. The development of central areolar choroidal dystrophy. Graefes Arch. Clin. Exp. Ophthalmol. 234, 87–93 (1996).
Ferry, A. P., Llovera, I. & Shafer, D. M. Central areolar choroidal dystrophy. Arch. Ophthalmol. 88, 39 (1972).
Gass, J. M. D. Stereoscopic Atlas of Macular Diseases. Diagnosis and Treatment, 3rd edn. (The C.V. Mosby Company, Saint-Louis, 1987).
Reig, C., Serra, A., Gean, E., Vidal, M., Arumí, J., De la Calzada, M. D. et al. A point mutation in the RDS-peripherin gene in a Spanish family with central areolar choroidal dystrophy. Ophthalmic. Genet. 16, 39–44 (1995).
Sorsby, A. Choroidal angiosclerosis with special reference to its hereditary character. Br. J. Ophthalmol. 23, 433–444 (1939).
Piguet, B., Heon, E., Munier, F. L., Grounauer, P. A., Niemeyer, G., Butler, N. et al. Full characterization of the maculopathy associated with an Arg-172-Trp mutation in the RDS/peripherin gene. Ophthalmic Genet. 17, 175–186 (1996).
Hoyng, C. B., Heutink, P., Testers, L., Pinckers, A., Deutman, A. F. & Oostra, B. A. Autosomal dominant central areolar choroidal dystrophy caused by a mutation in codon 142 in the peripherin/RDS gene. Am. J. Ophthalmol. 121, 623–629 (1996).
Downes, S. M., Fitzke, F. W., Holder, G. E., Payne, A. M., Bessant, D. A., Bhattacharya, S. S. et al. Clinical features of codon 172 RDS macular dystrophy. Arch. Ophthalmol. 117, 1373–1383 (1999).
Yanagihashi, S., Nakazawa, M., Kurotaki, J., Sato, M., Miyagawa, Y. & Ohguro, H. Autosomal dominant central areolar choroidal dystrophy and a novel Arg195Leu mutation in the peripherin/RDS gene. Arch. Ophthalmol. 121, 1458–1461 (2003).
Lotery, A. J., Ennis, K. T., Silvestri, G., Nicholl, S., McGibbon, D., Collins, A. D. et al. Localisation of a gene for central areolar choroidal dystrophy to chromosome 17p. Hum. Mol. Genet. 5, 705–708 (1996).
Hughes, A. E., Lotery, A. J. & Silvestri, G. Fine localisation of the gene for central areolar choroidal dystrophy on chromosome 17p. J. Med. Genet. 35, 770–772 (1998).
Lichanska, A. M., McGibbon, D., Silvestri, G. & Hughes, A. E. A physical and expression map of the D17S1810-D17S1353 region spanning the central areolar choroidal dystrophy locus. Cytogenet. Cell Genet. 93, 43–47 (2001).
Wigginton, J. E. & Abecasis, G. R. PEDSTATS: descriptive statistics, graphics and quality assessment for gene mapping data. Bioinformatics 21, 3445–3447 (2005).
Sambrook, J., Fritsh, E. F. & Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd edn. (Cold Spring Laboratory Press, Cold spring Harbor, New York, 1989).
Charfeddine, C., Mokni, M., Ben Mousli, R., Elkares, R., Bouchlaka, C., Boubaker, S. et al. A novel missense mutation in the gene encoding SLURP-1 in patients with Mal de Meleda from northern Tunisia. Br. J. Dermatol. 149, 1108–1115 (2003).
Abecasis, G. R., Cherny, S. S., Cookson, W. O. & Cardon, L. R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30, 97–101 (2002).
Klevering, B. J., van Driel, M., van Hogerwou, A. J., van De Pol, D. J., Deutman, A. F., Pinckers, A. J. et al. Central areolar choroidal dystrophy associated with dominantly inherited drusen. Br. J. Ophthalmol. 86, 91–96 (2002).
Boon, C. J., Klevering, B. J., Cremers, F. P., Zonneveld-Vrieling, M. N., Theelen, T., Hollander, A. I. et al. Central areolar choroidal dystrophy. Ophthalmology 116, 771–782 (2009).
Gass, J. D. Dystrophies des cônes in Atlas stéréoscopique des affections maculaires, 3rd edn. 264–266 (Flammarion Paris, 1991).
Chopdar, A. A variant of central areolar choroidal dystrophy. Ophthalmic Paediatrics Genetics 14, 151–154 (1993).
Hoyng, C. B., van Rijn, P. M. & Deutman, A. F. Central areolar choroidal dystrophy and slowly progressive sensorineural hearing loss. Acta. Ophthalmol. Scand. 74, 639–641 (1996).
Acknowledgements
We thank the patients and their family members for their cooperation in this study. We are grateful to Pr Christian Kubisch for the fruitful discussion. This work was supported by the Tunisian Ministry of Higher Education, Scientific and Technological Research and the Ministry of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website (http://www.nature.com/jhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Ouechtati, F., Belhadj Tahar, O., Mhenni, A. et al. Central areolar choroidal dystrophy associated with inherited drusen in a multigeneration Tunisian family: exclusion of the PRPH2 gene and the 17p13 locus. J Hum Genet 54, 589–594 (2009). https://doi.org/10.1038/jhg.2009.82
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.82
