
Abstract
The Himalayan mountain range has played a dual role in shaping the genetic landscape of the region by (1) delineating east–west migrations including the Silk Road and (2) restricting human dispersals, especially from the Indian subcontinent into the Tibetan plateau. In this study, 15 hypervariable autosomal STR loci were employed to evaluate the genetic relationships of three populations from Nepal (Kathmandu, Newar and Tamang) and a general collection from Tibet. These Himalayan groups were compared to geographically targeted worldwide populations as well as Tibeto-Burman (TB) speaking groups from Northeast India. Our results suggest a Northeast Asian origin for the Himalayan populations with subsequent gene flow from South Asia into the Kathmandu valley and the Newar population, corroborating a previous Y-chromosome study. In contrast, Tamang and Tibet exhibit limited genetic contributions from South Asia, possibly due to the orographic obstacle presented by the Himalayan massif. The TB groups from Northeast India are genetically distinct compared to their counterparts from the Himalayas probably resulting from prolonged isolation and/or founder effects.
Similar content being viewed by others


Introduction
The Himalayan mountain range encompasses an unparalleled landscape featuring some of the planet's highest peaks providing a formidable barrier separating the Tibetan plateau from the Indian subcontinent.1 The region also constitutes an ethnic and linguistic contact zone as it marks a crossroad of three different religions (Islam, Hinduism and Buddhism) and two language families (Indo-European and Sino-Tibetan), with Sino-Tibetan partitioning into the Chinese and Tibeto-Burman (TB) subfamilies. Speakers of the latter represent the majority of the Himalayan inhabitants whereas the Indo-Europeans are restricted to the south of the cordillera.
The geo-political partitioning of TB-speaking groups include Tibet, Nepal, Northeast India, Bhutan, Burma and parts of Southeast Asia.2 Two opposing views on the origin of TB populations have been proposed. Both models invoke demic diffusion for the dispersal of the language subfamily. Su et al.3, 4 postulate the upper and middle Yellow River basin as the ancestral source of TB people. In contrast, van Driem5, 6 argues for the Yangtse River in Sichuan province as the homeland of TB speakers, with subsequent northward migrations to the fertile plains of the Yellow River valley where Neolithic civilizations flourished.
Our recent study on Y-chromosomal diversity in the Himalayan populations of Nepal and Tibet revealed high frequencies of East Asian-specific haplogroup O3a5-M134, congruent with earlier reports on TB groups,4, 7, 8, 9 suggesting a common ancestry for this language subfamily.1 This investigation also revealed elevated levels of South Asian-derived Y-haplogroup R1a1-M198 in the Nepalese populations of Kathmandu and Newar, indicating significant genetic influences from the Indian subcontinent, whereas the Himalayas served as a barrier to gene flow from the south into the Tibetan plateau.1 These findings are consistent with a recent mtDNA study,10 which reported reciprocal maternal gene flow between India and Nepal.
On the other hand, previous Y-chromosome studies characterizing the Tibetan people4, 11, 12 argue for Central Asian genetic contributions to account for the presence of Asian-specific YAP (Y Alu polymorphism) chromosomes (D-M174) in the plateau. Haplogroup D-M174 lineages are found at appreciable frequencies in the Andaman Islands of the Indian Ocean13 but only minimally in all East Asian populations with the exception of Japan.1, 4, 11, 12, 14, 15 Distinctive D-M174 subclades are observed in the Andaman Islands (D-M174*) and Japan (D2-M55), whereas Tibetans exhibit haplogroups D1-M15 and D3-P47.
In addition to the demic diffusion scenarios portrayed by linguistic and genetic studies,4, 5, 6 Aldenderfer and Yinong16 have proposed an acculturation mechanism to explain the spread of Neolithic civilizations throughout northwestern Tibet. The authors deem it unlikely that agricultural migrants from relatively low elevations in the eastern plateau acclimated to the alpine and desert steppes of the Changtang (northwestern Tibet) considering the physiological stresses imposed by the high altitudes. As a result, they find it more plausible for cultural and not migration to be the mechanism leading to the nomadic pastoralism currently practiced in the region.
Northeast Indian populations have been extensively studied due to their strategic location as well as their socio-cultural and linguistic diversity.9, 17, 18, 19, 20, 21, 22 This region, bordered by the Himalayas to the north and the Bay of Bengal in the south, connects the Indian subcontinent to East Asia. In a recent autosomal microsatellite study, the genetic affinities present among TB groups from Northeast India21 have been ascribed to geographic contiguity rather than linguistic affiliation. The same group suggested that, given its geographical proximity, Burma most likely provided the source population for the Manipur and Tripura territories, whereas those from the northern region (Arunachal Pradesh, Mizoram and Sikkim) have been populated primarily by Tibetans.21 Unfortunately, the study did not incorporate both probable parental source populations to assess their phylogenetic relationships to the Northeast Indian groups. Other studies based on Bhutanese and Nepalese Y-STR profiles revealed isolation and drift among these Himalayan collections, with a pronounced effect of drift observed in Bhutan. Nevertheless, haplotype sharing indicates possible gene flow between them or from a common source population.23, 24
Autosomal STRs are selectively neutral, hypervariable markers that vary solely on the basis of mutation and drift.25 They are distributed throughout the genome and biparentally inherited, as opposed to the uniparental Y-chromosome and mtDNA polymorphisms, providing a more holistic representation of the evolutionary and migratory events that have shaped modern populations. These attributes endow them with the high resolution necessary to assess phylogenetic relationships among closely related human groups.26, 27, 28, 29
Although several studies related to the Tibetan population utilizing autosomal markers have been previously published,30, 31, 32, 33, 34, 35 the current project is the first of its kind to perform comprehensive phylogenetic analyses in a statistically significant sample size. A set of 15 autosomal STR loci was employed to characterize three populations from Nepal (Newar, Tamang and Kathmandu) as well as the general populace from Tibet. These collections were compared to previously published geographically and ethnically targeted groups from East Asia, South Asia and Southwest Asia in an attempt to establish genetic relationships among them.
This study aims to provide information on the genetic origins of Tibetan and Nepalese populations. Our results suggest that these Himalayan populations are of Northeast Asian ancestry and that the Kathmandu and Newari collections have been significantly influenced by subsequent gene flow from South Asia. However, the genetic contribution of this latter region to the Tamang and Tibet is negligible, probably due to the physical barrier presented by the Himalayan massif. It is noteworthy that the three Nepalese populations (Kathmandu, Newar and Tamang) display strong genetic differentiation despite sharing close geographic confines, underscoring the need for studies of anthropologically well-defined ethnic groups. A second objective is to assess the phylogenetic relationships between our Himalayan populations and Northeast Indian TB peoples. Contrary to previously published Y-chromosome data,9 our work indicates that Northeast Indians are genetically distinct from the Himalayan and East Asian collections, a disparity that may be attributable to male founder effects.
Materials and methods
Sample collection and DNA isolation
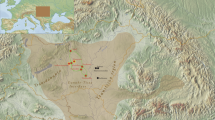
Blood samples were collected with informed consent from 341 unrelated individuals from Tibet (153) and three populations from Nepal (188), namely Tamang (45), Newar (66) and Kathmandu (77). The ancestry of every person was recorded for a minimum of two generations. With the exception of the people of Kathmandu, who speak an Indo-European language (Nepali), members of the three other collections are TB speakers. The geographic locations of the populations are shown in Figure 1. Sample collections were performed in accordance with the ethical guidelines put forth by the institutions involved in this study. DNA was extracted by the standard phenol–chloroform and ethanol precipitation method as described in Antunez de Mayolo et al.36 and stored at −80 °C.

Geographic locations of the Himalayan and reference populations examined in this study.
DNA amplification and STR genotyping
DNA samples were amplified at 15 autosomal STR loci in a multiplex reaction using the AmpFlSTR Identifiler kit.37 PCR amplification was performed in a GeneAmp PCR System 9600 thermocycler37 following the cycling conditions described elsewhere.26, 27 Amplicons were separated by multi-capillary electrophoresis in an ABI Prism 3100 Genetic Analyzer37 and ABI program Genescan 500 LIZ was utilized as an internal size standard. The Genescan 3.7 program was employed to determine the fragment sizes and alleles were designated by comparison to an allelic ladder from the manufacturer using Genotyper 3.7 NT software.
Statistical and phylogenetic analyses
STR allelic frequencies were calculated utilizing the GENEPOP program in the web software v3.4.38 The Arlequin software package version 2.00039 was employed to estimate observed and expected heterozygosities (Ho and He, respectively) as well as to determine gene diversity indices (GDI). Several parameters of population genetics interest, including power of discrimination (PD), matching probability (MP), polymorphic information content (PIC), power of exclusion (PE) and typical paternity index (TPI) were obtained with the PowerStats program version 1.2.40, 41, 42
Allelic frequencies of the four collections in the current study as well as the reference populations (Table 1) were utilized to generate Neighbor Joining (NJ) dendrograms based on Nei's genetic distances for two different datasets involving 15 and 13 STR loci, respectively, using the PHYLIP v3.6 program.55 D2S1338 and D19S433 were excluded for the 13 loci analyses (from the 15 loci set) as the data for these loci were not reported for all reference populations. The robustness of the phylogenetic relationships established by the NJ tree was assessed using bootstrap analysis with 1000 reiterations. Correspondence Analyses (CA) were also performed with the two sets of 15 and 13 STR loci with the NTSYSpc-2.02i software.56
Pairwise comparisons were conducted to ascertain genetic affinity between a given pair of populations with the Carmody program's G test57 involving 100 000 simulations. The Bonferroni correction was applied to account for the possibility of Type I errors resulting for the multiplicity of the tests performed. The DISPAN program58 was employed to compute inter-, intra- and total population variance (Gst, Hs and Ht, respectively) at the level of 13 STR loci. The populations analyzed were classified based on their geographical distributions as displayed in Table 1. In addition, DISPAN was utilized to calculate the average heterozygosity for each population.
Admixture estimations were generated by a Weighted Least Squares (WLS) method25, 59 using the Statistical Package for the Social Sciences (SPSS) 14.0 software. The WLS model calculates admixture proportions based on the following equation:

where pih is the frequency of the ith allele in the hybrid population, pij represents the frequency of the ith allele in the jth parental group (j=1, …., J), μj is the proportionate contribution of the jth parental gene pool to the hybrid population, and ∑jμj=1 (where, 0⩽μj⩽1).
Parental groups were partitioned according to geographical location as follows: Northeast Asia (Japan, Korea and Shaanxi), Southeast Asia (Chao Shan, Thailand, Malaysia and Philippines), South Asia (Pakistan, Punjab and Bangladesh) and Southwest Asia (Afghanistan, Iran and Iraq). Their contributions to the five Himalayan collections (Tibet, Tamang, Newar, Kathmandu and Bhutan) were examined across the 15 loci.
Results
Intra-population diversity
The allelic frequency distributions for Tibet, Tamang, Newar and Kathmandu are listed in Supplementary Tables 1 to 4. Combined Matching Probability (CMP), Combined Power of Exclusion (CPE), Combined Power of Discrimination (CPD) and Average GDI are provided in Supplementary Table 5. Loci D7S820 and THO1 in Kathmandu and D13S317 in Tibet were found to depart from Hardy–Weinberg equilibrium (HWE) expectations, however, these deviations were rendered statistically insignificant after applying the Bonferroni correction (α=0.05/15=0.0033). The highest GDI in Tibet is found at the FGA locus (0.8792) whereas the maximum values for this parameter are observed at D18S51 (0.8597) and D2S1338 (0.8854 and 0.8823) in Tamang, Newar and Kathmandu, respectively. Intra-population variance (Hs) values are presented in Supplementary Table 6. The Southwest Asians possess the highest intra-population variance (0.7801) followed by the Himalayan (0.7771) and Southeast Asian (0.7767) collections, all of which are higher than the all-population group (0.7739), whereas the Northeast Asian assemblage exhibits the lowest value (0.7713).
Inter-population diversity: 15 STR loci analyses
Phylogenetic analyses based on allelic frequencies were performed to investigate the genetic relationships between the four Himalayan collections and other geographically targeted populations using the set of 15 STR loci. Four separate aggregates are evident in the CA plot (Figure 2): both Northeast Asian and Himalayan clusters (except for Kathmandu and Newar) in the upper right quadrant, the Southeast Asian assemblage in the lower right portion and the South Asia/Southwest Asia cluster on the left side of the graph. Kathmandu plots intermediate between the South/Southwest Asian group and the Himalayan and Northeast Asian collections. Newar is an outlier near the perimeter of the upper left quadrant whereas Tibet and Tamang map close to each other within the Himalayan assemblage. In the NJ phylogram (Figure 4), the Himalayan groups occupy an intermediate position between the East and South/Southwest Asian populations. Tibet, Tamang and Bhutan form a separate clade whereas Newar branches between Kathmandu and the general population from Nepal.

Correspondence Analysis (CA) employing 15 STR loci.
G-test results at the 15 STR loci level are presented in Table 2. Only the Kathmandu and Bangladesh pair displayed non-significant genetic differences (G=186.0376, P=0.1139) from a total of 171 pairwise population comparisons at α=0.05. However, after applying the Bonferroni correction (α=0.05/171=0.0002924) several comparisons yielded additional statistically insignificant co-relationships (Table 2). Pairwise analyses using the 13 STR loci dataset generated similar results to those using 15 loci with the exception of an additional statistically insignificant populations pair (Iran/Punjab) (data not shown).
Admixture proportions were generated for the five Himalayan populations, namely Tibet, Tamang, Newar, Kathmandu and Bhutan using Northeast Asia, Southeast Asia, South Asia and Southwest Asia as parental populations (Table 3). When examining the genetic contributions from the parental groups, Northeast Asia arises as the major donor to Tibet (63.4%), Tamang (59.7%) and Newar (44.7%) and also contributes considerable proportions of the Bhutan (41.1%) and Kathmandu (22.3%) gene pools. Bhutan (44.3%), on the other hand is mainly influenced by Southeast Asia whereas relatively low inputs from the same region were observed in Tamang (29.0%), Tibet (21.1%) and Kathmandu (5.4%). In contrast, Kathmandu (63.3%) and Newar (41.5%) are primarily affected by the South Asian group, whereas the latter impacts the other Himalayan populations only minimally (Tibet 2.7%) or not at all (Tamang, 0.0% and Bhutan, 0.0%). All of the Himalayan collections experienced similar levels of gene flow from Southwest Asia (9–15%).
Inter-population diversity: 13 CODIS core STR loci analyses
To compare our Himalayan populations to TB groups from Northeast India and other Asian populations, allelic frequencies from the 13 CODIS STR loci shared among 25 collections were employed to perform additional phylogenetic analyses. Three well delineated groupings are apparent in the CA plot (Figure 3): the Northeast Indian assemblage (except for the Naga) in the lower right quadrant, the Southwest Asians with some South Asian populations in the lower left portion and the Himalayan/Northeast/Southeast Asian aggregate (except for Kathmandu and Newar) in the upper half of the graph. The NJ dendrogram (Figure 5) supports the information provided by the CA plot with the exception of three Himalayan groups (Tibet, Tamang and Bhutan) which form a sister clade with the Northeast Indian populations. Interestingly, populations within this Himalayan and Northeast Indian cluster belong to the same TB language subfamily.

Correspondence Analysis (CA) using 13 STR loci.
Inter- and total population variance (Gst and Ht, respectively) are reported in Supplementary Table 6. South Asia displays the highest Gst (0.0222) while also sharing the greatest Ht values (0.7894) with the All-population group. In contrast, Northeast Asia exhibits the lowest Gst and Ht diversities (0.0036 and 0.7740, respectively). The Himalayan populations possess intermediate Gst and Ht values (0.0120 and 0.7811, respectively).
Discussion
In this study, 15 autosomal STR loci were typed in three Nepalese populations (Tamang, Newar and Kathmandu) and in a general collection from Tibet to investigate their genetic ancestry and phylogenetic relationships to other TB communities and worldwide populations. This work improves on an earlier study54 by examining ethnic groups from Nepal that are anthropologically well-defined. Furthermore, this report complements previous Y-chromosome data1 thereby providing a comprehensive analysis of the genetic diversity in the Himalayas.
On average, the Himalayan populations (Newar, Kathmandu, Tamang and Tibet) possess 126 alleles whereas Northeast, Southeast and the South Asian populations examined in this study average 151, 140 and 137 allelic variants, respectively. The lower genetic diversity of the Himalayan collections is also reflected in their relatively reduced average heterozygosity values (0.7862) when compared to those of Northeast Asia (0.7890), Southeast Asia (0.7907) and South Asia (0.8009). Of the four, Tamang displays the lowest genetic variance reflected in both its average gene diversity index value (0.7632) and average heterozygosity value (0.7702), a characteristic echoed in its limited Y-chromosomal diversity.1
The inter-population diversity (Gst) (Supplementary Table 6) among the Himalayan cluster (0.0120) is considerably higher than in Northeast (0.0036), Southwest (0.0053) and Southeast Asian (0.0068) populations but is lower than among the South Asian collections (0.0222) and the All-populations group (0.0196). The concurrence of limited heterozygosity and elevated Gst value in the Himalayan populations could be results of multiple genetic sources, genetic drift and/or founder effect. In addition, selective pressure and adaptation to high altitudes, possibly in combination with inbreeding and patrilocality, could also promote homozygosity and genetic differences among the populations. The relatively high Gst value observed among the South Asians is also reflected in the disperse partitioning of the Northeast Indian populations in the CA plot (Figure 3) and may reflect known socio-cultural and genetic barriers. High degree of genetic differentiation resulting from various source populations may account for the greater variance among Himalayan populations in comparison to the Northeast, Southeast and Southwest Asian assemblages.
It is noteworthy that all three Nepalese groups not only exhibit considerable genetic diversity among themselves but also with respect to the general population of Nepal (Figures 2 and 3). Kathmandu plots closer to the South Asian groups while still maintaining genetic proximity with the Himalayan and Northeast Asian clusters (Figures 2 and 3). Pairwise G-test comparisons between Kathmandu and Bangladesh (G=186.0376, P=0.1139) and Kathmandu and Pakistan (G=213.5188, P=0.0014) are statistically insignificant, after application of Bonferroni correction, indicating some degree of genetic homogeneity between these South Asian and Nepalese populations. Although Newar segregates from all other populations at the periphery of the upper left quadrant in the 15 loci CA graph (Figure 2), its location is intermediate between the South Asia and other Himalayan collections in the NJ phylogram (Figure 4). It is possible that the Newari population (an ethnic minority) has been subject to genetic isolation and drift generating this divergent phylogenetic profile. Conversely, genetic impacts from distinct source populations may have contributed to Newar's unique genetic partitioning. The latter possibility is also reflected in a previous study19 based on 11 genetic markers (blood groups, red cell enzyme and serum proteins) in which Newaris were found to cluster with two geographically distant Manipuri groups from Northeast India in the NJ tree rather than with Tamang and Gurung, both of Nepalese descent.

Neighbor Joining (NJ) tree based on Nei's genetic distances generated using allele frequencies from the 15 STR loci. The numbers at the nodes represent bootstrap values estimated from 1000 replications.
The admixture profiles (Table 3) reveal a substantial proportion of South Asian genes (63.3%) in Kathmandu as compared to Northeast Asia (22.3%), supporting the relationships illustrated by both the G test and CA plot. Newar, on the other hand, experienced similar contributions from both regions (44.7 vs 41.5%), possibly accounting for its equidistant positioning to these groups of populations in the CA plot (Figure 3) and the NJ tree (Figure 4). The South Asian influence in the Nepalese populations may be associated with the entry of Aryans from the Indian plains who introduced the Indo-European language (Nepali) in the Kathmandu valley. A recent mtDNA study10 also reported reciprocal maternal gene flow between North India and Nepal. These results are consistent with the elevated levels of Y-haplogroup R in Newar (62.1%) and Kathmandu (46.7%), which are most likely derived from South Asia, particularly North India, given the geographic vicinity as well as the historical and socio-cultural affinities shared between these two neighboring regions.1 In contrast, Tamang and Tibet exhibit minimal percentages of Y-haplogroup R (8.8 and 2.5%, respectively), indicating that the Himalayas served as a formidable orographic barrier to gene flow from the south.1 Findings from the current investigation lend support to the aforementioned statement as admixture results reveal a null contribution from South Asia to both Tamang and Bhutan and only a minor genetic impact onto the Tibetan collection (2.7%). The absence of the South Asian signature in the gene pools of Tamang and Bhutan may be the result of geographic isolation and/or founder effects from another source population(s).
Close genetic ties have been reported between the Tamang and Tibet.1 It is likely that Tamangs are descendants of Tibetans who migrated south and settled in the southern region of the Himalayan range.1 This affinity is reflected in both CA plots (Figures 2 and 3) and NJ dendrograms (Figures 4 and 5). The Tibetan connection to the Tamang is also evident in their shared cultural and religious practices. The partitioning of these two populations with Bhutan and their proximity to the general collection from Nepal (Figures 2, 3, 4 and 5) may be associated with Neolithic migrants carrying Y-haplogroup O3a5-M134, an East Asian-specific marker, shared among TB populations.1, 3, 4, 9, 60 The Himalayan populations, with the exception of Newar and Kathmandu, segregate close to the Northeast Asian cluster in agreement with the admixture analyses results (Table 3). Northeast Asia is the major contributor to both Tibet (63.4%) and Tamang (59.7%) whereas Newar (44.7%) and Bhutan (41.1%) received equivalent percentages, followed by Kathmandu (22.3%). These results corroborate studies indicating a shared common ancestry between Tibet and the Northeast Asian collections of Japan and Korea by a variety of marker systems, including classical,61, 62 autosomal,63 Y-chromosome1, 12, 64, 65 and mtDNA.12, 64, 66, 67

Neighbor Joining (NJ) tree based on Nei's genetic distances generated using allele frequencies from the 13 CODIS core STR loci. The numbers at the nodes represent bootstrap values estimated from 1000 replications.
More than half of the Tibetan men possess the YAP polymorphic Alu insertion in their Y-chromosome which is believed to have originated in Central Asia,1, 4, 11, 14 although its source remains highly debated.64, 68, 69 In this study, however, given the lack of representative Central Asian populations due to the paucity of the data available from the region, no clear connections were made between Tibet and its possible Central Asian genetic contributors. Afghanistan is the sole Central Asian collection included in the analyses and appears to make no contributions to any of the Himalayan groups except for a minor influence in Kathmandu (12.9%).
To evaluate the genetic relationships between the Himalayan collections and the neighboring TB-speaking populations at the regional level, six Northeast Indian TB groups were included in the phylogenetic and statistical analyses performed using the 13 core CODIS STR loci. These Northeast Indian TB groups map distantly from both the Himalayan and East Asian populations in the CA graph (Figure 3), inconsistent with previous Y-chromosome and mtDNA studies which report a high degree of genetic homogeneity between Himalayan and Northeast Indian TB groups.3, 4, 9, 70 The discrepancy observed between Y-chromosome and microsatellite polymorphisms in the Northeast Indian TB groups may be explained by a male founder effect from Northeast Asia and their subsequent genetic isolation for an extended period of time following their arrival.9
Altogether, our results suggest a Northeast Asian ancestry for the Himalayan populations with subsequent genetic admixture in Kathmandu and Newar populations from South Asia. South Asian influences in Tibet and Tamang are negligible most likely due to the natural barrier presented by the Himalayas.1 Tamang, Tibet and Bhutan display close genetic affiliations in all analyses possibly indicating a shared common ancestry. The biparental markers examined in this study reveal unique genetic profiles for the Northeast Indian TB groups, which are distinct from their Himalayan counterparts implying limited gene flow, geographic isolation and/or founder effects.
References
Gayden, T., Cadenas, A. M., Regueiro, M., Singh, N. B., Zhivotovsky, L. A., Underhill, P. A. et al. The Himalayas as a directional barrier to gene flow. Am. J. Hum. Genet. 80, 884–894 (2007).
Gordon, R. G. Ethnologue: Languages of the World, 15th edn (SIL International, Dallas, 2005).
Su, B., Xiao, J., Underhill, P., Deka, R., Zhang, W., Akey, J. et al. Y-Chromosome evidence for a northward migration of modern humans into Eastern Asia during the last Ice Age. Am. J. Hum. Genet. 65, 1718–1724 (1999).
Su, B., Xiao, C., Deka, R., Seielstad, M. T., Kangwanpong, D., Xiao, J. et al. Y chromosome haplotypes reveal prehistorical migrations to the Himalayas. Hum. Genet. 107, 582–590 (2000).
van Driem, G. L. in Tibeto-Burman Phylogeny and Prehistory: Languages, Material Culture, and Genes (eds Bellwood, P. & Renfrew, C.) 233–249 (MacDonald Institute for Archaeological Research, University of Cambridge, United Kingdom, 2002).
van Driem, G. L. in The Peopling of East Asia: Putting Together Archaeology, Linguistics and Genetics (eds Sagart L., Blench. R. & Sanches-Mazas, A.) 81–106 (Routledge Curzon, Taylor and Francis group, London, 2005).
Sengupta, S., Zhivotovsky, L. A., King, R., Mehdi, S. Q., Edmonds, C. A., Chow, C. E. et al. Polarity and temporality of high-resolution y-chromosome distributions in India identify both indigenous and exogenous expansions and reveal minor genetic influence of Central Asian pastoralists. Am. J. Hum. Genet. 78, 202–221 (2006).
Sahoo, S., Singh, A., Himabindu, G., Banerjee, J., Sitalaximi, T., Gaikwad, S. et al. A prehistory of Indian Y chromosomes: evaluating demic diffusion scenarios. Proc. Natl Acad. Sci. USA 103, 843–848 (2006).
Cordaux, R., Weiss, G., Saha, N. & Stoneking, M. The northeast Indian passageway: a barrier or corridor for human migrations? Mol. Biol. Evol. 21, 1525–1533 (2004).
Thangaraj, K., Chaubey, G., Kivisild, T., Selvi Rani, D., Singh, V. K., Ismail, T. et al. Maternal footprints of Southeast Asians in North India. Hum. Hered. 66, 1–9 (2008).
Qian, Y., Qian, B., Su, B., Yu, J., Ke, Y., Chu, Z. et al. Multiple origins of Tibetan Y chromosomes. Hum. Genet. 106, 453–454 (2000).
Hammer, M. F., Karafet, T. M., Park, H., Omoto, K., Harihara, S., Stoneking, M. et al. Dual origins of the Japanese: common ground for hunter-gatherer and farmer Y chromosomes. J. Hum. Genet. 51, 47–58 (2006).
Thangaraj, K., Singh, L., Reddy, A. G., Rao, V. R., Sehgal, S. C., Underhill, P. A. et al. Genetic affinities of the Andaman Islanders, a vanishing human population. Curr. Biol. 13, 86–93 (2003).
Hammer, M. F., Spurdle, A. B., Karafet, T., Bonner, M. R., Wood, E. T., Novelletto, A. et al. The geographic distribution of human Y chromosome variation. Genetics 145, 787–805 (1997).
Ding, Y. C., Wooding, S., Harpending, H. C., Chi, H. C., Li, H. P., Fu, Y. X. et al. Population structure and history in East Asia. Proc. Natl Acad. Sci. USA 97, 14003–14006 (2000).
Aldenderfer, M. & Yinong, Z. The prehistory of the Tibetan plateau to the seventh century A.D.: perspectives and research from China and the West since 1950. J. World. Prehist. 18, 1–55 (2004).
Mastana, S. S., Murry, B., Sachdeva, M. P., Das, K., Young, D., Das, M.K. et al. Genetic variation of 13 STR loci in the four endogamous tribal populations of Eastern India. Forensic. Sci. Int. 169, 266–273 (2007).
Maity, B., Nunga, S. C. & Kashyap, V. K. Genetic polymorphism revealed by 13 tetrameric and 2 pentameric STR loci in four Mongoloid tribal population. Forensic. Sci. Int. 132, 216–222 (2003).
Kumar, V., Basu, D. & Reddy, B. M. Genetic heterogeneity in northeastern India: reflection of Tribe-Caste continuum in the genetic structure. Am. J. Hum. Biol. 16, 334–345 (2004).
Krithika, S., Trivedi, R., Kashyap, V. K. & Vasulu, T. S. Genetic diversity at 15 microsatellite loci among the Adi Pasi population of Adi tribal cluster in Arunachal Pradesh, India. Leg. Med. (Tokyo) 7, 306–310 (2005).
Krithika, S., Trivedi, R., Kashyap, V. K., Bharati, P. & Vasulu, T. S. Antiquity, geographic contiguity and genetic affinity among Tibeto-Burman populations of India: a microsatellite study. Ann. Hum. Biol. 33, 26–42 (2006).
Kashyap, V. K., Chattopadhyay, P., Dutta, R. & Vasulu, T. S. Genetic structure and affinity among eight ethnic populations of Eastern India: based on 22 polymorphic DNA loci. Am. J. Hum. Biol. 16, 311–327 (2004).
Parkin, E. J., Kraayenbrink, T., van Driem, G. L., Tshering Of Gaselo, K., de Knijff, P. & Jobling, M. A. 26-Locus Y-STR typing in a Bhutanese population sample. Forensic. Sci. Int. 161, 1–7 (2006).
Parkin, E. J., Kraayenbrink, T., Opgenort, J. R., van Driem, G. L., Tuladhar, N. M., de Knijff, P. et al. Diversity of 26-locus Y-STR haplotypes in a Nepalese population sample: isolation and drift in the Himalayas. Forensic. Sci. Int. 166, 176–181 (2007).
Perez-Miranda, A. M., Alfonso-Sanchez, M. A., Pena, J. A. & Herrera, R. J. Qatari DNA variation at a crossroad of human migrations. Hum. Hered. 61, 67–79 (2006).
Shepard, E. M. & Herrera, R. J. Iranian STR variation at the fringes of biogeographical demarcation. Forensic. Sci. Int. 158, 140–148 (2006a).
Shepard, E. M. & Herrera, R. J. Genetic encapsulation among Near Eastern populations. J. Hum. Genet. 51, 467–476 (2006b).
Shepard, E. M., Chow, R. A., Suafo’a, E., Addison, D., Perez-Miranda, A. M., Garcia-Bertrand, R. L. et al. Autosomal STR variation in five Austronesian populations. Hum. Biol. 77, 825–851 (2005).
Rowold, D. J. & Herrera, R. J. Inferring recent human phylogenies using forensic STR technology. Forensic. Sci. Int. 133, 260–265 (2003).
Ying, B., Liu, T., Chen, G., Zhao, Z., Fan, H. & Wen, F. Genetic profile of two Tibetan populations from China by analysis of 15 STR loci. Hum. Biol. 78, 757–770 (2006).
Yan, J., Shen, C., Li, Y., Yu, X., Xiong, X., Mu, H. et al. Genetic analysis of 15 STR loci on Chinese Tibetan in Qinghai province. Forensic. Sci. Int. 169, e3–e6 (2007).
Ota, M., Droma, Y., Basnyat, B., Katsuyama, Y., Asamura, H., Sakai, H. et al. Allele frequencies for 15 STR loci in Tibetan populations from Nepal. Forensic. Sci. Int. 169, 234–238 (2007).
Kang, L. L., Zhao, J., Liu, K. & Li, X. Allele frequencies of 15 STR loci of Tibetan lived in Tibet Lassa. Forensic. Sci. Int. 168, 236–240 (2007).
Kang, L., Yuan, D., Yang, F., Liu, K. & Za, X. Genetic polymorphisms of 15 STR loci in two Tibetan populations from Tibet Changdu and Naqu, China. Forensic. Sci. Int. 169, 239–243 (2007).
Kang, L. & Li, S. Allele frequencies of 15 STR loci of Luoba ethnic group in Tibet (Southwestern China). Forensic. Sci. Int. 155, 219–221 (2005).
Antunez-de-Mayolo, G., Antunez-de-Mayolo, A., Antunez-de-Mayolo, P., Papiha, S. S., Hammer, M., Yunis, J. J. et al. Phylogenetics of worldwide human populations as determined by polymorphic Alu insertions. Electrophoresis 23, 3346–3356 (2002).
Applied Biosystems. AmpFlSTR Identifiler PCR Amplification Kit User's Manual (Applied Biosystems, Foster City, CA, USA, 2001).
Raymon, M. & Rousset, F. Genepop (version 1.2): population genetics software for exact tests and ecumenicism. J. Hered. 86, 248–249 (1995).
Schneider, S., Kueffer, J. M., Roessli, D. & Excoffier, L. Arlequin v. 2.000: A Software for Population Genetic Analysis (Genetics and Biometry Laboratory, University of Geneva, Geneva, 2000).
Jones, D. A. Blood samples: probability of discrimination. J. Forensic. Sci. Soc. 12, 355–359 (1972).
Brenner, C. H. & Morris, J. W Proceedings for the International Symposium on Human Identification (Promega Corporation, Madison, 1990).
Tereba, A. Profiles in DNA (Promega Corporation, Madison, 1999).
Hashiyada, M., Itakura, Y., Nagashima, T., Nata, M. & Funayama, M. Polymorphism of 17 STRs by multiplex analysis in Japanese population. Forensic. Sci. Int. 133, 250–253 (2003).
Kim, Y. L., Hwang, J. Y., Kim, Y. J., Lee, S., Chung, N. G., Goh, H. G. et al. Allele frequencies of 15 STR loci using AmpF/STR Identifiler kit in a Korean population. Forensic. Sci. Int. 136, 92–95 (2003).
Wang, Z. Y., Yu, R. J., Wang, F., Li, X. S. & Jin, T. B. Genetic polymorphisms of 15 STR loci in Han population from Shaanxi (NW China). Forensic. Sci. Int. 147, 89–91 (2005).
Hu, S. P., Yu, X. J., Liu, J. W. & Cai, K. L. Analysis of STR polymorphisms in the Chao Shan population in South China. Forensic. Sci. Int. 147, 93–95 (2005).
Seah, L. H., Jeevan, N. H., Othman, M. I., Jaya, P., Ooi, Y. S., Wong, P. C. et al. STR data for the AmpFlSTR Identifiler loci in three ethnic groups (Malay, Chinese, Indian) of the Malaysian population. Forensic. Sci. Int. 138, 134–137 (2003).
De Ungria, M. C. A., Roby, R. K., Tabbada, K. A., Rao-Coticone, S., Tan, M. M. & Hernandez, K. N. Allele frequencies of 19 STR loci in a Philippine population generated using AmpFlSTR multiplex and ALF singleplex systems. Forensic. Sci. Int. 152, 281–284 (2005).
Rerkamnuaychoke, B., Rinthachai, T., Shotivaranon, J., Jomsawat, U., Siriboonpiputtana, T., Chaiatchanarat, K. et al. Thai population data on 15 tetrameric STR loci—D8S1179, D21S11, D7S820, CSF1PO, D3S1358, TH01, D13S317, D16S539, D2S1338, D19S433, vWA, TPOX, D18S51, D5S818 and FGA. Forensic. Sci. Int. 158, 234–237 (2006).
Berti, A., Barni, F., Virgili, A., Iacovacci, G., Franchi, C., Rapone, C. et al. Autosomal STR frequencies in Afghanistan population. J. Forensic. Sci. 50, 1494–1496 (2005).
Barni, F., Berti, A., Pianese, A., Boccellino, A., Miller, M. P., Caperna, A. et al. Allele frequencies of 15 autosomal STR loci in the Iraq population comparisons to other populations from the middle-eastern. Forensic. Sci. Int. 167, 87–92 (2007).
Dobashi, Y., Kido, A., Fujitani, N., Susukida, R., Hara, M. & Oya, M. STR data for the AmpFlSTR Identifiler loci in Bangladeshi and Indonesian populations. Leg. Med. 7, 222–226 (2005).
Kraaijenbrink, T., van Driem, G. L., Tshering of Gaselo, K. & de Knijff, P. Allele frequency distribution for 21 autosomal STR loci in Bhutan. Forensic. Sci. Int. 170, 68–72 (2007a).
Kraaijenbrink, T., van Driem, G. L., Opgenort, J. R. M. L., Tuladhar, N. M. & de Knijff, P. Allele frequency distribution for 21 autosomal STR loci in Nepal. Forensic. Sci. Int. 168, 227–231 (2007b).
Felsentein, J. Phylogeny Inference Package (PHYLIP) Version 3.6a3 (Department of Genetics, University of Washington, Seattle, 2002).
Rohlf, F. NTSYSpc (Exter Publishing, Setauket, NY, 2002).
Carmody, G. G-test (Carleton University, Ottawa, 1990).
Ota, T. DISPAN: Genetic Distance and Phylogenetic Analysis (Institute of Molecular Evolutionary Genetics, Pennsylvania State University, Pennsylvania, 1993).
Long, J. C., Williams, R. C., McAuley, J. E., Medis, R., Partel, R., Tregellas, W. M. et al. Genetic variation in Arizona Mexican Americans: estimation and interpretation of admixture proportions. Am. J. Phys. Anthropol. 84, 141–157 (1991).
Black, M. L., Wise, C. A., Wang, W. & Bittles, A. H. Combining genetics and population history in the study of ethnic diversity in the People's Republic of China. Hum. Biol. 78, 277–293 (2006).
Cavalli-Sforza, L. L., Menozzi, P. & Piazza, A. The History and Geography of Human Genes (Princeton University Press, Princeton, NJ, USA, 1994).
Nei, M. & Roychoudhury, A. K. Evolutionary relationships of human populations on a global scale. Mol. Biol. Evol. 10, 927–943 (1993).
Chu, J. Y., Huang, W., Kuang, S. Q., Wang, J. M., Xu, J. J. & Chu, Z. T. Genetic relationship of populations in China. Proc. Natl Acad. Sci. USA 95, 11763–11768 (1998).
Tajima, A., Hayami, M., Tokunaga, K., Juji, T., Matsuo, M., Marzuki, S. et al. Genetic origins of the Ainu inferred from combined DNA analyses of maternal and paternal lineages. J. Hum. Genet. 49, 187–193 (2004).
Tajima, A., Pan, I. H., Fucharoen, G., Fucharoen, S., Matsuo, M., Tokunaga, K. et al. Three major lineages of Asian Y chromosomes: implications for the peopling of east and southeast Asia. Hum. Genet. 110, 80–88 (2002).
Tanaka, M., Cabrera, V. M., Gonzalez, A. M., Larruga, J. M., Takeyasu, T., Fuku, N. et al. Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome. Res. 14, 1832–1850 (2004).
Torroni, A., Miller, J. A., Moore, L. G., Zamudio, S., Zhuang, J., Droma, T. et al. Mitochondrial DNA analysis in Tibet: implications for the origin of the Tibetan population and its adaptation to high altitude. Am. J. Phys. Anthropol. 93, 189–199 (1994).
Jin, L. & Su, B. Natives or immigrants: modern human origin in east Asia. Nat. Rev. Genet. 1, 126–133 (2000).
Underhill, P. A. in The Peopling of East Asia: Putting Together Archaeology, Linguistics, and Genetics (eds. L. Sagart., R. Blench. & A. Sanchez-Mazas.) 301–319 (Routledge Curzon, Taylor and Francis group, London, 2004).
Wen, B., Xie, X., Gao, S., Li, H., Shi, H., Song, X. et al. Analyses of genetic structure of Tibeto-Burman populations reveals sex-biased admixture in southern Tibeto-Burmans. Am. J. Hum. Genet. 74, 856–865 (2004).
Acknowledgements
We gratefully acknowledge Diane J Rowold and Javier R Luis for their constructive criticism of the article, and Charlie Haz and Michael Solier for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website (http://www.nature.com/jhg)
Rights and permissions
About this article
Cite this article
Gayden, T., Mirabal, S., Cadenas, A. et al. Genetic insights into the origins of Tibeto-Burman populations in the Himalayas. J Hum Genet 54, 216–223 (2009). https://doi.org/10.1038/jhg.2009.14
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.14
Keywords
This article is cited by
-
Sequence variations, flanking region mutations, and allele frequency at 31 autosomal STRs in the central Indian population by next generation sequencing (NGS)
Scientific Reports (2021)
-
Allelic frequency database of 15 polymorphic autosomal STRs in the Malayalam-speaking population of Kerala, India
International Journal of Legal Medicine (2020)
-
Estimating genetic polymorphism in Bhuiyan population of eastern India using 20 autosomal STR loci
International Journal of Legal Medicine (2020)
-
Genetic variation (population database) at 20 autosomal STR loci in the population of Rajasthan (north-western India)
International Journal of Legal Medicine (2020)
-
Differentiated demographic histories and local adaptations between Sherpas and Tibetans
Genome Biology (2017)
