
Abstract
The aim of this work was to study the mutations within ATP7B in Egyptian children with Wilson disease and to evaluate any potential correlation between genotype and phenotype in this cohort. The study consisted of 48 children with Wilson disease from 32 independent families. The 21 exons of the ATP7B gene were amplified in a thermal cycler. Direct sequencing of the amplified polymerase chain reaction (PCR) products was performed by cycle sequencing using fluorescent dye terminators in an automatic ABI sequencer. Thirty-one different mutations in 96 chromosomes were detected (19 missense, three nonsense, seven frameshift deletions, and two splice-site mutations). Of these, 12 mutations have not been previously reported. The p.N1270S, p.C703Y, IVS18-2A > G, p.R1319X, c.2304-2305insC, and p.H1069Q were present in 7.8%, 6.2%, 6.2%, 6.2%, 4.7%, and 4.7%, respectively, of studied chromosomes in independent families. One patient was homozygous for both p.N1270S and p.T1434M mutations. Frameshift and nonsense mutations were found in 50% of patients with disease onset ≤8 years compared with only 26% in patients with onset >8 years. Despite mutation heterogeneity in Egyptian children, genotype–phenotype correlation analysis seems to be promising in this population, as many patients carry homozygous mutations, a situation that mandates a larger-scale population screening to identify the carrier rate in this community.
Similar content being viewed by others

Introduction
Wilson disease (WD) is an autosomal recessive disorder of copper transport characterized by decreased hepatobiliary copper excretion and reduced copper incorporation into ceruloplasmin (Danks 1989). ATP7B, the gene mutated in WD, consists of 21 exons and encodes a 1,465 amino acid protein representing a copper transporting P-type adenosine triphosphatase (ATPase) (Thomas et al. 1995).
Mutation screening in WD patients has led to the detection of at least 300 disease-specific mutations (University of Alberta Department of Medical Genetics 2002). The published data suggest that some mutations appear to be population specific, whereas others are common to many populations. The spectrum of mutations and their clinical consequences has not previously been studied in the Egyptian population. In addition, mutational studies in pediatric WD are still limited in the literature.
The aim of this work was to study mutations within ATP7B in Egyptian children with WD and to evaluate any potential correlation between genotype and phenotype in this Egyptian cohort.
Patients and methods
The study included 48 children (28 boys and 20 girls) from 32 independent families with WD who presented to Yassin Abdelghaffar Charity Center for Liver Disease and Research (a major tertiary referral center for liver diseases in Egypt) and the hepatology clinic, Children’s Hospital, Ain Shams University. Children referred from different parts of Egypt (37.5% of families from lower Egypt, 33.3% from upper Egypt, 20.8% from Cairo and Giza, and 8.3% from Sinaa). For each family, full medical history of all members including those with the disease was recorded and included family pedigree analysis. Screening of suspected family members was performed by clinical examination, ceruloplasmin oxidase activity in serum, and liver function tests, and was confirmed by mutational analysis.
The diagnosis of WD was established in the presence of at least two out of three of the following criteria: low ceruloplasmin level <20 mg/dl or the presence of Kayser–Fleischer rings by slit-lamp examination and/or hepatic copper content of 250 μg/g dry weight liver tissue in the presence of hepatic or neurological manifestations consistent with WD (Sternlieb 1990). Liver involvement was ascertained based on clinical evaluation, liver function tests, ultrasonography, and liver biopsy when necessary. Neuropsychiatric involvement was based on clinical evaluation and brain magnetic resonance imaging (MRI) in individual cases.
ATPB mutation analysis
Genomic DNA was extracted from whole venous blood collected in ethylenediaminetetraacetate (EDTA) using standard procedures from the 48 patients and 113 unrelated healthy Egyptian subjects. The 21 exons of the WD gene were amplified in a thermal cycler (Biometra T3 Thermocycler, Göttingen, Germany) as described elsewhere (Waldenström et al. 1996). Direct sequencing of the amplified polymerase chain reaction (PCR) products was performed by cycle sequencing using fluorescent dye terminators in an automatic sequencer (Applied Biosystems, Darmstadt, Germany). Mutations were quoted according to the guidelines from http://www.HGVS.org/mutnomen/ using the reference sequence with the GenBank accession number NM_00053.1. The nucleotide +1 is the A of the ATG-translation initiation codon. The ATG-translation initiation codon is also the first codon.
RFLP analysis and allele-specific PCR
The novel mutations modifying endonuclease restriction sites were studied by specific restriction fragment length polymorphism (RFLP) analysis. The novel mutations that did not modify any known restriction sites were analyzed by allele-specific PCR (Table 1).
Results
Sequencing of the ATP7B gene revealed 31 different mutations in 96 chromosomes (19 missense, three nonsense, seven frameshift deletions, and two splice-site mutations). Of these, 12 mutations had not been previously reported (novel mutations). Novel mutations included five missense, two nonsense, four frameshift deletions, and one splice-site mutation (Table 1). They were confirmed using RFLP analysis or allele-specific oligohybridization analysis. None of the mutations were found in the 226 chromosomes of 113 healthy subjects.
Consanguinity was present in 75% of families. Affected family members shared the same genotype. In some instances, the same genotype was shared by more than one family (Table 2). One patient was homozygous for both p.N1270S and p.T1434M mutations.
From the 32 families studied, 32 children (index or symptomatic patients) presented with clinical manifestations suggestive of WD, and 16 sibs were diagnosed by screening family members of index patients. Whereas the index patients presented with hepatic or/and neurological symptoms, all screened children had only hepatic manifestations (Table 3). Index patients characteristics in relation to mutations are described in Table 4. In the first group (children who where <8 years of age when they had their symptoms), most patients were boys, and all had hepatic symptoms only with no neurological manifestations, which was statistically significant (p value 0.03). Frameshift and nonsense mutations (indicative of severe disease) were the responsible mutation in 50% of patients compared with 26% in children who presented at >8 years of age.
The diversity of mutations limited our genotype–phenotype association analysis; however, several observations were noted: frameshift mutations affected more than one third of children with hepatic phenotype, and in 46% of these cases, the child presented with subacute or acute hepatic failure. Splice-site mutation seems to result in severe disease, as patients carrying this type of mutation either presented with decompensated cirrhosis (two patients) or they later develop neurological manifestations (three patients). Frameshift mutations were significantly higher in patients with hepatic phenotype, whereas splice-site mutations were significantly higher in patients with neurological phenotype (Tables 5, 6).
There was a tendency of mutations in the transmembrane domains and ATP loop to result in early onset of disease (≤8 years). Also, mutations of the ATP loop resulted in hepatic symptoms with absence of neurological manifestations, whereas ATP hinge mutations resulted in hepatic failure in half of patients, and transmembrane and copper-binding domain mutations were associated with neurological manifestations (Tables 7, 8).
Discussion
To date, at least 300 different mutations have been identified in WD chromosomes. Some mutations appear to be population specific [e.g., p.H1069Q in populations of European origin (Tanzi et al. 1993); p.R778L in Asian populations (Thomas et al. 1995)]. Unlike the European and Chinese populations, we found no single predominant mutation in Egyptian children. However, p.N1270S mutation was present in 7.8%; p.C703Y, IVS18-2A > G, and p.R1319X were each present in 6.2%; and p.H1069Q and c.2304-2305insC in 4.7% of studied chromosomes in independent families. A probable explanation of this diversity is that the Egyptian population is very heterogeneous with respect to ethnicity, and it may also indicate a high carrier rate for WD in this community.
In this study, children with disease onset <8 years of age were mostly boys, which may be explained by the cultural philosophy of better care for boys. Mean age of appearance of neurological symptoms did not differ from that of hepatic symptoms (9.8 compared with 10.9 years); furthermore, onset of neurological symptoms was much earlier than that reported in the literature (Machado et al. 2006).
Genotype–phenotype correlation analysis was limited in this study due to diversity of the mutations detected, but it was found that frameshift and nonsense mutations (a possible predictor of severe phenotype) were found in 50% of children with age of onset <8 years compared with only 26% of patients with age of onset >10 years. It was also observed that frameshift mutations affected more than one third of children with hepatic phenotype, and in about 46% of these cases, the child presented with hepatic failure. On the other hand, splice-site mutation resulted in hepatic failure, and children having this mutation all developed neurological manifestations. Also, when the mutation affected the ATP loop, it resulted in early presentation of disease and hepatic symptoms with absence of neurological manifestations, whereas ATP hinge mutations resulted in hepatic failure in half the patients, and transmembrane mutations resulted in the appearance of neurological manifestations. All these observations require larger-scale patient analysis to confirm their significance.
It should be noted that because all patients were referred from a pediatric hepatology clinic, a sample bias might have occurred. It was of interest to note that even in the same family member with the same genotype, children had different phenotypes. In one family carrying the homozygous p.E396X mutation, the index patient had neurological manifestation only at the age of 18 years (onset was 15 years), his younger sister had both hepatic and neurological manifestation at the age of 17, and the youngest brother had only hepatic manifestations until the writing of this study (20 years). In another family (homozygous IVS18-2A > G mutation), the index patient presented with fulminant hepatic failure at the age of 8 years, whereas his brother had only neurological manifestations, which began at the age of 10 years and continued without hepatic manifestations till this writing (15 years). This suggests that other factors (outside the ATPB7 gene or environmental factors) affect the disease phenotype (Takeshita et al. 2002).
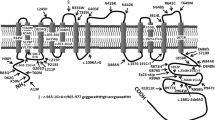
As Egypt is considered an Arab country in the Mediterranean region, we compared this study with other studies performed in Mediterranean or Arab populations. In the study of Loudianos et al. (1999) on Mediterranean patients with WD, ten out of 19 of the novel mutations found were localized on exons 14, 16, and 19, which belong to the group of exons previously found to be the site of many frequent, as well as rare, WD-causing mutations. The genetic analysis of 56 Saudi patients with WD revealed that 50% of them had mutations in three exons (8, 19, and 21) of the ATP7B gene. Mutations in exon 19 and 21 were unique for Saudi patients (Takeshita et al. 2002). A novel deletion mutation, c.4193delC, in exon 21 of the ATP7B gene mutation, appears to be unique to Saudi patients and is found frequently in this ethnic group (Al Jumah et al. 2004; Majumdar et al. 2000, 2003). This is in contrast to our study, in which the novel mutations detected were randomly distributed all over the ATP7B gene, including exons (2, 4, 5, 7, 11, 13, 15, 17–21; Fig. 1).

Distribution of new mutations on the ATP7B gene
Again, the majority of mutations detected in this study were found in the homozygous state (42 patients from 26 independent families), whereas in the study of Loudianos et al. (1999), most mutations detected were in the compound heterozygous state, and in only four cases, homozygosity was present. This may be explained by the high percentage of consanguinity in our study (75%) compared with consanguinity in the Egyptian population (32–35%).
Although homozygous mutations were found in 42 patients from 26 independent families, six of those patients from six different families were the result of nonconsanguineous mating. Also heterozygous mutations were found in six patients, two were the result of consanguineous mating. This unexpected finding, together with the finding of one patient homozygous for both p.N1270S and p.T1434M mutations, necessitates a larger-scale population study for the carrier frequency and the origins of these mutations in different families in this community.
Conclusion
The mutational spectrum of ATP7B among Egyptian children presenting with WD is very heterogenous, which mandates a larger-scale population screening to identify the carrier rate in this community. Frameshift and nonsense mutations may result in earlier presentation of the disease at childhood. Despite mutation heterogeneity in Egyptian patients, genotype–phenotype correlation analysis seems to be promising in this population, as many patients carry homozygous mutations.
References
Al Jumah M, Majumdar R, Al Rajeh S, Awada A, Al Zaben A, Al Traif I, Al Jumah AR, Rehana ZA (2004) Clinical and genetic study of 56 Saudi Wilson disease patients: identification of Saudi-specific mutations. Eur J Neurol 11:121–124
Danks DM (1989) Disorders of copper transport. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic basis of inherited diseases, 6th edn. McGraw-Hill, New York, pp 1416–1422
Loudianos G, Dessi V, Lovicu M, Angius A, Altunatus B, Giacchino R, Marazzi M, Marcellini M, Sartorelli MR, Sturniolo GC, Kocak N, Yuce A, Akar N, Pirastu M, Cao A (1999) 19 Novel mutations descent with Wilson disease: identification of mutation analysis in patients of Mediterranean. J Med Genet 36:833–836
Machado A, Chien HF, Deguti MM, Cançado E, Azevedo RS, Scaff M, Barbosa ER (2006) Neurological manifestations in Wilson’s disease: report of 119 cases. Mov Disord 21:2192–2196
Majumdar R, Al Jumah M, Al Rajeh S, Fraser M, Al Zaben A, Awada A, Al Traif I, Paterson M (2000) A novel deletion mutation in the carboxyl terminus of the copper-transporting ATPase gene causes Wilson disease. J Neurol Sci 179:140–143
Majumdar R, Al Jumah M, Fraser M (2003) 4193delC, a common mutation causing Wilson’s disease in Saudi Arabia: rapid molecular screening of patients and carriers. J Clin Pathol Mol Pathol 56:302–304
Sternlieb I (1990) Perspectives on Wilson’s disease. Hepatology 12:1234–1239
Takeshita Y, Shimizu N, Yamaguchi Y, Nakazono H, Saitou M, Fujikawa Y, Aoki T (2002) Two families with Wilson disease in which siblings showed different phenotypes. J Hum Genet 47:543–547
Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, Romano DM, Parano E, Pavone L, Brzustowicz LM, Devoto M, Peppercorn J, Bush AI, Sternlieb I, Pirastu M, Gusella JF, Evgrafov O, Penchaszadeh GK, Honig B, Edelman IS, Soares MB, Scheinberg IH, Gilliam TC (1993) The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet 5:344–350
Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW (1995) The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet 9:210–217
University of Alberta Department of Medical Genetics. http://www.uofamedical-genetics.org, accessed 30 September, 2002
Waldenström E, Lagerkvist A, Dahlman T, Westermark K, Landegren U (1996) Efficient detection of mutations in Wilson disease by manifold sequencing. Genomics 37:303–309
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Abdelghaffar, T.Y., Elsayed, S.M., Elsobky, E. et al. Mutational analysis of ATP7B gene in Egyptian children with Wilson disease: 12 novel mutations. J Hum Genet 53, 681 (2008). https://doi.org/10.1007/s10038-008-0298-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10038-008-0298-7
Keywords
This article is cited by
-
Wilson’s disease clinic at the Assiut Liver Center in Egypt: a real well-established step on the way
Egyptian Liver Journal (2022)
-
Mutation analysis of the ATP7B gene and genotype–phenotype correlation in Chinese patients with Wilson disease
BMC Gastroenterology (2021)
-
ATP7B variant penetrance explains differences between genetic and clinical prevalence estimates for Wilson disease
Human Genetics (2020)
-
Novel compound heterozygote mutations in the ATP7B gene in an Iranian family with Wilson disease: a case report
Journal of Medical Case Reports (2018)
-
The six metal binding domains in human copper transporter, ATP7B: molecular biophysics and disease-causing mutations
BioMetals (2017)
