
Abstract
Wilson disease (WD), an autosomal recessive disorder of copper transport, is the most common inherited liver disorder in Hong Kong Chinese. This was the first local study to elucidate the molecular basis and establish an effective DNA-based diagnostic protocol. The ATP7B genes of 65 patients were amplified by polymerase chain reaction (PCR) and sequenced. Haplotype analysis was performed using D13S301, D13S314, and D13S316. The p.L770L/p.R778L status in 660 subjects was determined to estimate WD prevalence. Allele age of p.R778L was determined by the smallest homozygosity region between D13S301 and D13S270. We identified 42 different mutations with 17 being novel. p.R778L (17.3%) was the most prevalent. Exons 2, 8, 12, 13, and 16 harbored 70% mutations. Thirty-two haplotypes were associated with WD chromosomes. The estimated prevalence rate was 1 in 5,400. Three out of 660 normal subjects had p.L770L/p.R778L. In the remaining 657 individuals, neither p.L770L nor p.R778L was found. We characterized a Hong Kong Chinese-specific ATP7B mutation spectrum with great genetic diversity. Exons 2, 8, 12, 13, and 16 should be screened first. The perfect linkage disequilibrium suggested that p.R778L and its private polymorphism p.L770L originated from a single ancestor. This East-Asian-specific mutation p.R778L/p.L770L is aged at least 5,500 years.
Similar content being viewed by others
Introduction
Wilson disease (WD, MIM # 277900) is an autosomal recessive disorder of copper transport leading to systemic copper accumulation and hepatic and lenticular damage. The worldwide prevalence was reported to be 1 in 30,000 with a carrier rate of 1 in 90 (Scheinberg and Sternlieb 1984). In our experience, WD is the most common inherited liver disease in Hong Kong Han Chinese. The mutation spectrum of the ATP7B gene is population specific even within East Asia (Ferenci 2006; Kenney and Cox 2007; Lee et al. 2000; Park et al. 2007; Shimizu et al. 1999; Wan et al. 2006). Therefore, data from other areas cannot be directly applicable for diagnostic purposes. We examined the genotypes and haplotypes of the ATP7B mutant alleles in 76 WD patients from 65 unrelated Hong Kong Han Chinese families. This was also the first study using a genetic approach to estimate the WD prevalence in Hong Kong Han Chinese. In particular, we utilized the p.R778L, which is the most common East Asia-specific mutation, to study the prehistoric migration of ancient Han Chinese in East Asia.
Materials and methods
Subjects
Sixty-five unrelated WD probands together with 11 presymptomatic siblings were recruited for the study. All were ethnic Han Chinese originating from Guangdong province. Consanguinity was reported in only one family. Diagnosis of WD was based on at least two of the followings: the presence of Kayser–Fleischer (KF) rings by slit lamp examination, typical neurological symptoms, and/or low serum ceruloplasmin levels (<0.20 g/L). In patients without KF rings and/or with normal serum ceruloplasmin, diagnosis was based on other laboratory parameters suggestive of impaired copper metabolism [elevated 24-h urinary copper excretion (>1.0 μmol/day) and/or hepatic copper content >250 μg/g dry weight] in the absence of cholestasis.
Analysis of the ATP7B gene
Genomic DNA was extracted from peripheral blood samples using a QIAamp Blood Kit (Qiagen, Hilden, Germany) after informed consent was obtained. The study was approved by our institutional ethnics committee. The coding exons and the flanking introns of the ATP7B gene were amplified by polymerase chain reaction (PCR). The PCR primers were carefully designed to avoid allele dropout (Lam and Mak 2006). We used one touch-down PCR approach for all exons: 94°C for 12 min, ten cycles of 94°C for 30 s, 66°C with an decrement of temperature by 1°C for each subsequent cycle for 30 s, 72°C for 45 s, 30 cycles of 94°C for 30s, 60°C for 30 s, 72°C for 45 s, and a final extension at 72°C for 8 min. The reaction mixture of a final volume of 25 μl contained 1× PCR buffer II (Applied Biosystems, Foster City, CA), 2.0 mM magnesium chloride, 200 μM dNTP, 1 μM of each primer, 0.625 units AmpliGold Taq polymerase (Applied Biosystems), and 100 ng DNA template.
Denaturing high performance liquid chromatography
We performed a heteroduplex analysis by denaturing high-performance liquid chromatography (DHPLC) on a WAVE DHPLC instrument (Transgenomic Inc., San Jose, CA). The stationary phase consists of 2-μm non-porous alkylated poly-styrene-divinylbenzene particles packed into a 50 × 4.6-mm ID DNASep column (Transgenomic Inc.). The PCR products were heated, denatured, and then cooled slowly to form a complete duplex. Ten microlitre samples were loaded, and the eluted DNA fragments were detected with ultraviolet absorption at wavelength 260 nm. The correct temperature for mutation scanning was determined by the WAVE utility software based on the wild-type DNA sequence.
Sequence analysis
Exons showing the presence of heteroduplexes by DHPLC and patient samples with inconclusive DHPLC chromatograms were sequenced directly. PCR products were purified by Microspin S-300 HR columns (GE Healthcare, Uppsala, Sweden), and both strands were sequenced using their amplification primers and BigDye–Deoxy terminator cycle sequencing reagents, according to the manufacturer’s instructions (Applied Biosystems). Products of the sequencing reactions were purified using Auto-Seq G-50 columns (GE Healthcare). Purified sequencing fragments were separated by capillary electrophoresis and detected by laser-induced fluorescence on an ABI Prism 3100 genetic analyzer.
Haplotype analysis
Three microsatellite markers (D13S314, D13S301, and D13S316) flanking the WD locus were used (Petrukhin et al. 1993; Thomas et al. 1993, 1994). The PCR was performed using previously published primers (Thomas et al. 1994) in 15-μl volume containing 9 μl ABI Prism True Allele PCR Premix (Applied Biosystems), 5 μM of each primer, and 60 ng DNA. The thermal condition was 94°C for 12 min, 35 cycles of 94°C for 15 s, 55°C for 20 s, 72°C for 30 s, and a final extension at 72°C for 40 min. The PCR products were diluted 1:10 with deionized water. The injection mix included 0.5 μl Genescan 400HD size standard (Applied Biosystems), 12 μl deionized formamide, and 1 μl diluted PCR products, and was denatured at 95°C for 5 min and quickly chilled on ice. The samples were electrophoresed on the ABI Prism 3100 genetic analyzer (Applied Biosystems), and data were analyzed with GeneMapper software version 3.0 (Applied Biosystems). The allele-size definitions of D13S314 and D13S316 were based on GDB:309065 and GDB:309089, respectively (http://www.gdb.org/gdb/). That of D13S301 was modified from previously published data (Chuang et al. 1996; Thomas et al. 1994), with additional allele sizes of 154 and 156 assigned as −1 and −2, respectively.
Allele age estimation of p.R778L and WD prevalence
The allele age of p.R778L was calculated based on the method of homozygosity mapping formula (1) proposed by Genin et al. (1998).
E(θn) is the expected length of the homozygosity region around the disease locus where no crossing over is expected to occur. In order to estimate the smallest homozygous region E(θ n) around the ATP7B gene on chromosome 13, we analyzed two additional microsatellite markers (D13S296 and D13S270) on 13 patients carrying p.R778L with complete family data (three homozygotes and eight heterozygotes). It is assumed that the disease allele p.R778L is rare enough to insure that an inbred affected has received two identical by descent copies, and not by chance. The PCR was performed using previously published primers (White et al. 1993) and the same thermal condition stated in the part of haplotype analysis. θ 0 is the length (in Morgans) of the whole chromosome where the disease locus maps. The θ 0 of chromosome 13 is 114 Morgans. The number of meioses in the inbred affected individuals was denoted by n.
We also recruited another 660 healthy Hong Kong Chinese subjects with informed consents and determined their p.L770L and p.R778L status for prevalence estimation. We adopted the mutation analysis approach published by Olivarez et al. (2001) and calculated the prevalence of WD in Hong Kong Chinese.
Results
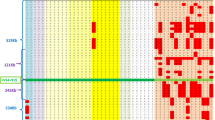
Forty-two different mutations across 16 exons were identified in 65 unrelated Hong Kong Chinese WD patients (Table 1). Seventeen novel mutations were found. Eleven were missense mutations, one nonsense, one deletion and four splicing mutations. All the novel missense mutations were non-conservative changes and were not found in 50 normal healthy subjects. The four most common mutations were c.2333G > T or p.R778L (17.3%), c.2975C > T or p.P992L (13.4%), c.3443T > C or p.I1148T (8.7%), and c.3532A > G or p.T1178A (5.5%). The mutation detection rate achieved was 97.6% (126 disease alleles found in 129 unrelated WD chromosomes) and was the highest among other major Asian studies (Okada et al. 2000; Park et al. 2007; Wan et al. 2006; Wu et al. 2001; Yoo 2002). In three patients, we found only one WD mutant allele after sequencing all the ATP7B exons and approximately 1 kb of each 5′ and 3′ untranslated region. In addition, we also identified 32 different microsatellite haplotypes associated with WD chromosomes in 33 probands with informative family data (Fig. 1).

Constructed microsatellite haplotypes and the associated mutations of the 33 WD families were shown. Numbers indicated the microsatellites of D13S314, D13S301, and D13S316 (from top to bottom), respectively. (ND: not done)
We also found four patients who carried both p.Q1142H and p.I1148T on the same allele and another mutation on the trans allele (i.e., p.A874V, p.T1178A, c.2304dupC, and IVS3 + 1G > T, respectively). Their parental data confirmed that p.Q1142H cosegregated in cis with p.I1148T. Both p.Q1142H and p.I1148T were previously described as disease-causing mutations (Loudianos et al. 1998a, 1999). We did not find either mutation in 50 normal healthy subjects. Patients carrying both p.Q1142H and p.I1148T should have their parental genotypes to confirm the compound heterozygosity on the homologous chromosomes or have all exons being sequenced because they may bear three mutations.
The most prevalent mutant allele p.R778L was found in 22 of 129 WD chromosomes. Moreover, p.R778L segregated with five different microsatellite haplotypes. The microsatellite haplotypes of D13S314, D13S301, and D13S316 were 13–3–7, 10–9–7, 10–6–7, 10–5–7, and 10–4–7 (Fig. 1.1-11). The three p.R778L homozygous patients carried all five haplotypes (Fig. 1.1–3). Nonetheless, all studied p.R778L alleles were in perfect linkage disequilibrium with a non-synonymous single nucleotide polymorphism c.2310C > G or p.L770L. We screened 660 healthy Hong Kong Han Chinese individuals using direct DNA sequencing and only three carriers with p.R778L and p.L770L were detected. In the remaining 657 individuals, neither p.L770L nor p.R778L was found. In addition, the carrier rate of p.R778L was 1 in 220 (allele frequency 0.0023). Because p.R778L accounted for one-sixth of all mutations in our study, the expected frequency of all WD mutant alleles was 0.0136. We adopted the mutation analysis approach published by Olivarez et al. (2001), and by Hardy–Weinberg equilibrium, the calculated prevalence of WD in Hong Kong Han Chinese was 1 in 5,400.
The physical position of the ATP7B gene is 51,404,806–51,483,631 bp on chromosome 13 (NM 001005918). Both D13S296 and D13S270 were situated at the downstream of ATP7B gene (Fig. 2). Among the three p.R778L homozygotes and eight p.R778L heterozygotes, all were homozygous for D13S296 with the same allele size of 128 bp, but heterozygous for D13S270. As a result, the smallest homozygous region around the ATP7B gene was flanked by D13S301 at the 5′ end and by D13S270 at the 3′ end. It was about 850 kb in length. This region encompassed a recombination cold spot (White et al. 1993). Using formula (1), we calculated that p.R778L may have originated 220 generations ago. Assuming an average generation of 25 years, this ancestral mutation was aged 5,500 years old.

The smallest homozygosity region around ATP7B gene on chromosome 13 was shown. Haplotypes of D13S314, D13S296, and D13S270 of the three p.R778L homozygotes (indicated with asterisks) and the eight heterozygotes were listed
Discussions
Our study was the first study to elucidate the genotype of ATP7B in Hong Kong Han Chinese WD patients and estimate the prevalence of WD in Hong Kong Chinese. We have carefully analyzed the genotype data of Hong Kong Han Chinese and expanded the mutation spectrum of the ATP7B gene with 17 novel mutations of total 42 different mutations in 65 unrelated WD patients. The genetic background is so far the most heterogeneous. In contrast, there were only 7 different mutations in 72 Northern Chinese patients (Liu et al. 2004) and 18 mutations among 84 patients (Wu et al. 2001), 10 mutations in 29 Taiwanese patients (Wan et al. 2006), 21 mutations in 41 Japanese patients (Okada et al. 2000), and 28 mutations in 120 Korean patients (Park et al. 2007). The rest of the mutations are highly variable. Most mutations lie on exons 8, 12, 13, 16, and 18 in Northern Chinese covering 60.5–74% mutations (Liu et al. 2004; Wu et al. 2001); exons 8, 12, 13, 16, and 18 in Taiwanese with coverage of 61.8% (Wan et al. 2006); exons 8, 11, and 18 in Koreans with coverage of 59.8–71.4% (Park et al. 2007; Yoo 2002); and exons 5, 8, 12, 13, and 18 in Japanese with coverage of 60.9–70% (Okada et al. 2000; Shimizu et al. 1999). We proposed a five-exon screening approach to Hong Kong Han Chinese. Exons 2, 8 12, 13, and 16 should be screened first. This five-exon approach can cover 70% of mutations.
The prevalence of WD in Hong Kong Han Chinese was 1 in 5,400. Our findings concurred with the prevalence reported in other East-Asian countries (Kim et al. 1998; Ohura et al. 1999; Yamaguchi et al. 1999) that WD seems more prevalent in East Asia than in western countries.
Intriguingly, p.R778L has been reported only in East-Asian populations and is absent in other ethnic groups. At the same amino acid 778 position, Caucasians have other amino acid substitutions. They were c.2332C > G or p.R778G in Turkish (Figus et al. 1995) and Greek patients (Loudianos et al. 1998a), c.2333G > A or p.R778Q in Greek (Loudianos et al. 1998a), and c.2332C>T or p.R778W in North American (Shah et al. 1997), Italian (Loudianos et al. 1998b), Sardinian (Loudianos et al. 1999) and British patients (Butler et al. 2001; Curtis et al. 1999), whereas these mutations have never been reported in Chinese patients, except p.R778Q (Lee et al. 2000).
Mutation p.R778L also has been observed on two other different microsatellite haplotypes (8–4–4 and 8–4–5.5) in Taiwanese (Chuang et al. 1996; Lee et al. 2000) and five microsatellite haplotypes (5–5–6, 5–7–4, 5–7–5, 5–7–5.5, and 5–7–7) in Japanese (Nanji et al. 1997). Nonetheless, despite the various associated microsatellite haplotypes, p.R778L in the Han population is always linked to the polymorphism p.L770L, which does not coexist with other p.R778 missense mutations and has not been reported in other ethnic groups. Moreover, the genetic distance of the three microsatellite markers used are approximately 1.5 cM (Thomas et al. 1995), whereas the p.R778L mutation (c.2333G>T) is only 23 bp away from p.L770L (c.2310C>G), thus the expected risk of recombination is negligible.
Additionally, p.L770L was not identified in 657 normal individuals without p.R778L. Therefore, we excluded p.R778L as a recurrent mutation, based on its perfect linkage disequilibrium with p.L770L. In addition, codon 770 is not a common mutation because no mutation or other polymorphism has been reported. In other words, p.L770L is a private and unique polymorphism of p.R778L. Based on the close proximity, the p.L770L and p.R778L mutations conceivably occurred synchronously. Combined with the evidence of associated multiple microsatellite haplotypes, p.R778L is probably the most ancient founder mutation of the ATP7B gene worldwide. The smallest homozygosity region around the ATP7B gene on chromosome 13 was analyzed to be 850 kb and, thus, the estimated allele age of p.R778L was at least 5,500 years. The estimated age was the most conservative approximate, because the homozygosity region can be narrowed down by using denser microsatellite markers and studying on a larger sample size.
Although p.R778L is still the most prevalent mutation in East Asia, our study reported the lowest allele frequency of p.R778L (17.3%) among Hong Kong Han Chinese patients: 2.6-fold lower than Han patients from Northern China (45.6%, χ 2 = 27.9, p < 0.0001) (Liu et al. 2004) and significantly lower than patients from Beijing, Fujian, Shandong, Jiangxi, Shanghai, and Zhejiang (37.7%, χ 2 = 14.2, p = 0.0002) (Wu et al. 2001), from Shandong, Hebei, Anhui, Sichuan, Jiangsu, and Liaoning (33.8%, χ 2 = 8.1, p = 0.0043) (Gu et al. 2003), from Korea (37.9%, χ 2 = 10.8, p = 0.0010) (Yoo 2002), and Taiwan (43.1%, χ 2 = 14.9, p = 0.0001) (Wan et al. 2006). Despite being two different ethnic populations, Korean and Northern Han Chinese shared almost an identical allele frequency of p.R778L in WD patients. The comparatively low occurrence of p.R778L/p.L770L in Hong Kong supported the hypothesis of a single southward migration of Han Chinese originating in the North and reflected an increasing pattern of genetic admixture going from north to south (Fig. 3). It is amazing that the pre-historic Han overcame geographical barriers like the Yangtze and the Yellow Rivers, and finally reached as far as Hong Kong, traveling more than 2,000 km southward. We speculated on how forces such as famine or war could have driven this migration. Some have speculated that WD carriers might have an increased resistance to infections and cancers (Chu and Hung 1993; Wilkinson et al. 1983). It is not certain whether the heterozygous state of p.R778L/p.L770L serves a distinct advantage in survival and reproduction, but its persistence related to its founder effect and unique migration pattern was demonstrated unambiguously.

Geographic distribution of common ATP7B mutations in East Asia. A proposed north-to-south prehistoric migration route of ancient Han carrying p.R778L/p.770L is indicated by the solid arrow
Based on immunoglobulin Gm allotypes of Chinese populations across the Yangtze River, Zhao and Lee hypothesized that the modern Chinese nation originated from two distinct populations, one population originating in the Yellow River valley and the other originating in the Yangtze River valley during early Neolithic times (3,000–7,000 years ago) (Zhao and Lee 1989). Studies on disease genes, such as phenylalanine hydroxylase (PAH) for phenylketonuria (Lo et al. 1993; Wang et al. 1991) and citrin (SLC25A13) for adult-onset type II citrillinemia (Lu et al. 2005), were consistent with the findings of Zhao and Lee. A founder mutation of classical hyperphenylalanemia, p.R413P, was mostly present in Chinese populations north of the Yangtze River (Wang et al. 1991), while a common mutation of SLC25A13, c.851_854del, only occurred in Chinese populations south of the Yangtze River (Lu et al. 2005). A complete description of early populations must allow, therefore, the possibility of admixture of Chinese populations across the Yangtze River. In this study, we provided solid evidence that an ancient Northern Han Chinese population carrying the founder mutation of WD, p.R778L/p.L770L, crossed the Yangtze and Yellow Rivers and migrated southward as far as Hong Kong.
In conclusion, we have delineated the mutation spectrum of the ATP7B gene in Hong Kong Han Chinese with 17 novel mutations identified. The genetic background of ATP7B mutant is very heterogeneous, and we recommend screening exons 2, 8, 12, 13, and 16, which cover 70% of mutations. Our study also underlined the utility of recessive disease-associated alleles in the study of human migrations. By merging this with the data in other studies from China, a north-to-south direction in the cline of p.R778L frequencies was discerned. The present population group inheriting p.R778L/p.L770L, now residing in Hong Kong, mainland China, Taiwan, Japan, and Korea, appeared to have descended from a single ancestor at least 5,500 years ago. Combined with the evidence of associated multiple microsatellite haplotypes, p.R778L is probably the most ancient founder mutation of the ATP7B gene worldwide. Since WD is a common genetic liver disease in China, with its large population size, determining the allele frequencies of p.R778L/p.L770L in WD patients in all the provinces in China can refine the prehistoric migration path of the world’s biggest ethnic population.
References
Butler P, McIntyre N, Mistry PK (2001) Molecular diagnosis of Wilson disease. Mol Genet Metab 72:223–230
Chu NS, Hung TP (1993) Geographic variations in Wilson’s disease. J Neurol Sci 117:1–7
Chuang LM, Wu HP, Jang MH, Wang TR, Sue WC, Lin BJ, Cox DW, Tai TY (1996) High frequency of two mutations in codon 778 in exon 8 of the ATP7B gene in Taiwanese families with Wilson disease. J Med Genet 33:521–523
Curtis D, Durkie M, Balac P, Sheard D, Goodeve A, Peake I, Quarrell O, Tanner S (1999) A study of Wilson disease mutations in Britain. Hum Mutat 14:304–311
Ferenci P (2006) Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing. Hum Genet 120:151–159
Figus A, Angius A, Loudianos G, Bertini C, Dessi V, Loi A, Deiana M, Lovicu M, Olla N, Sole G, De Virgiliis S, Lilliu F, Anna Maria Giulia F, Annamaria N, Raffaella G, Arrigo B, Maria M, Lucia Z, Nella AG, Matilde M, Antonello S, Angelo D, Cristiana B, Marcella D, Sinasi O, Nurten K, Nejat A, Selin K, Vahe M, Paskal C, Angelo B, Antonio C, Pirastu M (1995) Molecular pathology and haplotype analysis of Wilson disease in Mediterranean populations. Am J Hum Genet 57:1318–1324
Genin E, Todorov AA, Clerget-Darpoux F (1998) Optimization of genome search strategies for homozygosity mapping: influence of marker spacing on power and threshold criteria for identification of candidate regions. Ann Hum Genet 62:419–429
Gu YH, Kodama H, Du SL, Gu QJ, Sun HJ, Ushijima H (2003) Mutation spectrum and polymorphisms in ATP7B identified on direct sequencing of all exons in Chinese Han and Hui ethnic patients with Wilson’s disease. Clin Genet 64:479–484
Kenney SM, Cox DW (2007) Sequence variation database for the Wilson disease copper transporter, ATP7B. Hum Mutat
Kim EK, Yoo OJ, Song KY, Yoo HW, Choi SY, Cho SW, Hahn SH (1998) Identification of three novel mutations and a high frequency of the Arg778Leu mutation in Korean patients with Wilson disease. Hum Mutat 11:275–278
Lam CW, Mak CM (2006) Allele dropout in PCR-based diagnosis of Wilson disease: mechanisms and solutions. Clin Chem 52:517–520
Lee CC, Wu JY, Tsai FJ, Kodama H, Abe T, Yang CF, Tsai CH (2000) Molecular analysis of Wilson disease in Taiwan: identification of one novel mutation and evidence of haplotype-mutation association. J Hum Genet 45:275–279
Liu XQ, Zhang YF, Liu TT, Hsiao KJ, Zhang JM, Gu XF, Bao KR, Yu LH, Wang MX (2004) Correlation of ATP7B genotype with phenotype in Chinese patients with Wilson disease. World J Gastroenterol 10:590–593
Lo WH, Wang T, Eisensmith R, Woo SL (1993) Molecular basis of PKU in China. Chin Med Sci J 8:180–185
Loudianos G, Dessi V, Lovicu M, Angius A, Kanavakis E, Tzetis M, Kattamis C, Manolaki N, Vassiliki G, Karpathios T, Cao A, Pirastu M (1998a) Haplotype and mutation analysis in Greek patients with Wilson disease. Eur J Hum Genet 6:487–491
Loudianos G, Dessi V, Lovicu M, Angius A, Nurchi A, Sturniolo GC, Marcellini M, Zancan L, Bragetti P, Akar N, Yagci R, Vegnente A, Cao A, Pirastu M (1998b) Further delineation of the molecular pathology of Wilson disease in the Mediterranean population. Hum Mutat 12:89–94
Loudianos G, Dessi V, Lovicu M, Angius A, Altuntas B, Giacchino R, Marazzi M, Marcellini M, Sartorelli MR, Sturniolo GC, Kocak N, Yuce A, Akar N, Pirastu M, Cao A (1999) Mutation analysis in patients of Mediterranean descent with Wilson disease: identification of 19 novel mutations. J Med Genet 36:833–836
Lu YB, Kobayashi K, Ushikai M, Tabata A, Iijima M, Li MX, Lei L, Kawabe K, Taura S, Yang Y, Liu TT, Chiang SH, Hsiao KJ, Lau YL, Tsui LC, Lee DH, Saheki T (2005) Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency. J Hum Genet 50:338–346
Nanji MS, Nguyen VT, Kawasoe JH, Inui K, Endo F, Nakajima T, Anezaki T, Cox DW (1997) Haplotype and mutation analysis in Japanese patients with Wilson disease. Am J Hum Genet 60:1423–1429
Ohura T, Abukawa D, Shiraishi H, Yamaguchi A, Arashima S, Hiyamuta S, Tada K, Iinuma K (1999) Pilot study of screening for Wilson disease using dried blood spots obtained from children seen at outpatient clinics. J Inherit Metab Dis 22:74–80
Okada T, Shiono Y, Hayashi H, Satoh H, Sawada T, Suzuki A, Takeda Y, Yano M, Michitaka K, Onji M, Mabuchi H (2000) Mutational analysis of ATP7B and genotype–phenotype correlation in Japanese with Wilson’s disease. Hum Mutat 15:454–462
Olivarez L, Caggana M, Pass KA, Ferguson P, Brewer GJ (2001) Estimate of the frequency of Wilson’s disease in the US Caucasian population: a mutation analysis approach. Ann Hum Genet 65:459–463
Park S, Park JY, Kim GH, Choi JH, Kim KM, Kim JB, Yoo HW (2007) Identification of novel ATP7B gene mutations and their functional roles in Korean patients with Wilson disease. Hum Mutat 28(11):1108–1113
Petrukhin K, Fischer SG, Pirastu M, Tanzi RE, Chernov I, Devoto M, Brzustowicz LM, Cayanis E, Vitale E, Russo JJ, Matseoane D, Boukhgalter B, Wasco W, Figus AL, Loudianos J, Cao A, Sternlieb I, Evgrafov O, Parano E, Pavone L, Warburton D, Ott J, Penchaszadeh GK, Scheinberg IH, Gilliam TC (1993) Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat Genet 5:338–343
Scheinberg IH, Sternlieb I (1984) Wilson’s disease. Major Probl Intern Med 23:9–16
Shah AB, Chernov I, Zhang HT, Ross BM, Das K, Lutsenko S, Parano E, Pavone L, Evgrafov O, Ivanova-Smolenskaya IA, Anneren G, Westermark K, Urrutia FH, Penchaszadeh GK, Sternlieb I, Scheinberg IH, Gilliam TC, Petrukhin K (1997) Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype–phenotype correlation, and functional analyses. Am J Hum Genet 61:317–328
Shimizu N, Nakazono H, Takeshita Y, Ikeda C, Fujii H, Watanabe A, Yamaguchi Y, Hemmi H, Shimatake H, Aoki T (1999) Molecular analysis and diagnosis in Japanese patients with Wilson’s disease. Pediatr Int 41:409–413
Thomas GR, Bull PC, Roberts EA, Walshe JM, Cox DW (1994) Haplotype studies in Wilson disease. Am J Hum Genet 54:71–78
Thomas GR, Roberts EA, Rosales TO, Moroz SP, Lambert MA, Wong LT, Cox DW (1993) Allelic association and linkage studies in Wilson disease. Hum Mol Genet 2:1401–1405
Thomas GR, Roberts EA, Walshe JM, Cox DW (1995) Haplotypes and mutations in Wilson disease. Am J Hum Genet 56:1315–1319
Tsai CH, Tsai FJ, Wu JY, Chang JG, Lee CC, Lin SP, Yang CF, Jong YJ, Lo MC (1998) Mutation analysis of Wilson disease in Taiwan and description of six new mutations. Hum Mutat 12:370–376
Wan L, Tsai CH, Tsai Y, Hsu CM, Lee CC, Tsai FJ (2006) Mutation analysis of Taiwanese Wilson disease patients. Biochem Biophys Res Commun 345:734–738
Wang T, Okano Y, Eisensmith RC, Harvey ML, Lo WH, Huang SZ, Zeng YT, Yuan LF, Furuyama JI, Oura T (1991) Founder effect of a prevalent phenylketonuria mutation in the oriental population. Proc Natl Acad Sci USA 88:2146–2150
White A, Tomfohrde J, Stewart E, Barnes R, Le Paslier D, Weissenbach J, Cavalli-Sforza L, Farrer L, Bowcock A (1993) A 4.5-megabase yeast artificial chromosome contig from human chromosome 13q14.3 ordering 9 polymorphic microsatellites (22 sequence-tagged sites) tightly linked to the Wilson disease locus. Proc Natl Acad Sci USA 90:10105–10109
Wilkinson ML, Portmann B, Williams R (1983) Wilson’s disease and hepatocellular carcinoma: possible protective role of copper. Gut 24:767–771
Wu ZY, Wang N, Lin MT, Fang L, Murong SX, Yu L (2001) Mutation analysis and the correlation between genotype and phenotype of Arg778Leu mutation in Chinese patients with Wilson disease. Arch Neurol 58:971–976
Yamaguchi Y, Aoki T, Arashima S, Ooura T, Takada G, Kitagawa T, Shigematsu Y, Shimada M, Kobayashi M, Itou M, Endo F (1999) Mass screening for Wilson’s disease: results and recommendations. Pediatr Int 41:405–408
Yoo HW (2002) Identification of novel mutations and the three most common mutations in the human ATP7B gene of Korean patients with Wilson disease. Genet Med 4:43S–48S
Zhao TM, Lee TD (1989) Gm and Km allotypes in 74 Chinese populations: a hypothesis of the origin of the Chinese nation. Hum Genet 83:101–110
Acknowledgments
We would like to thank the patients and their families; without their cooperation, this work would not have been possible. We are indebted to our clinical colleagues at the different hospitals who collected patient blood samples and clinical information. The work described in this paper was fully supported by a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (project no. CUHK4084/02M).
Author information
Authors and Affiliations
Corresponding author
Additional information
Chloe M MAK and Ching-Wan LAM contributed equally to this paper.
An erratum to this article is available at http://dx.doi.org/10.1007/s10038-007-0244-0.
Rights and permissions
About this article
Cite this article
Mak, C.M., Lam, CW., Tam, S. et al. Mutational analysis of 65 Wilson disease patients in Hong Kong Chinese: Identification of 17 novel mutations and its genetic heterogeneity. J Hum Genet 53, 55–63 (2008). https://doi.org/10.1007/s10038-007-0218-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-007-0218-2
Keywords
This article is cited by
-
Molecular genetic diagnosis of Wilson disease by ARMS-PCR in a Pakistani family
Molecular Biology Reports (2018)
-
Genetic analysis of 55 northern Vietnamese patients with Wilson disease: seven novel mutations in ATP7B
Journal of Genetics (2017)
-
Wilson’s Disease in China
Neuroscience Bulletin (2017)
-
Mutational analysis of ATP7B in north Chinese patients with Wilson disease
Journal of Human Genetics (2013)
-
Isolated persistent elevation of alanine transaminase for early diagnosis of pre-symptomatic Wilson’s disease in Chinese children
World Journal of Pediatrics (2013)
