
Abstract
Keratoconus is a bilateral ectatic disorder characterized by the central thinning of corneal tissue leading to visual impairment. To investigate the possibility of visual system homeobox 1 (VSXI) as a candidate susceptibility gene for Korean patients with keratoconus, we performed a mutation screening of the VSXI gene in 249 unrelated patients with keratoconus and 208 control subjects without the ocular disorder. We found two heterozygous novel missense mutations in exon 2: N151S and G160V. The G160V mutation was identified in 13 keratoconus patients (5.3%), and the N151S mutation was found in only one keratoconus patient (0.4%). We also detected three synonymous polymorphisms and four intragenic polymorphisms. The IVS1-11*a allele was associated with a significantly increased risk of keratoconus in Korean patients [3.6 vs. 0.5%, p = 0.001, odds ratio (OR) = 7.76, 95% confidence interval (CI) 1.989–30.241). Other polymorphisms did not show an association with keratoconus risk. Our data is the first reported VSX1 mutation screening in Korean keratoconus patients. We detected two novel missense mutations and one intragenic polymorphism in the VSX1 gene, which show a strong statistical association with unrelated keratoconus patients. Consequently, our study suggests that VSX1 gene variants seem to be significant genetic variants for keratoconus predisposition in unrelated Korean patients.
Similar content being viewed by others
Introduction
Keratoconus (KTCN; OMIM 148300) is a noninflammatory, bilateral ectatic disorder characterized by central thinning of corneal tissue with myopia and astigmatism that leads to visual impairment (Krachmer et al. 1984). Histologically, thinning and scarring of corneal stroma, iron deposition in the epithelial basement membrane, disruption of Bowman’s layer, and eventual protrusion of the cornea can be observed (Nakamura et al. 2005; Sherwin and Brookes 2004). The prevalence of keratoconus is estimated to be 1 per 2,000 in the general population. The onset of keratoconus is in puberty, and the progression of the disease occurs through the third or fourth decade of life, when it usually stops (Karseras and Ruben 1976; Rabinowitz 1998).
The exact cause of keratoconus is unclear, although its pathogenesis may involve genetic, environmental, and behavioral factors. It may be associated with various factors, including atopy, eye rubbing, hard contact lenses, and numerous multisystemic genetic diseases, including Down syndrome, Leber’s congenital amaurosis, and Ehlers-Danlos syndrome (Bawazeer et al. 2000; Chwa et al. 2006; Elder 1994; Flanders et al. 1984; Kenney and Brown 2003; Kuming and Joffe 1977; Rabinowitz 1998; Shapiro and France 1985). Family-based and twin studies have shown that genetic factors in particular play a significant role in the development of keratoconus and that there is a high concordance rate for keratoconus in monozygotic twins (Edwards et al. 2001; McMahon et al. 1999; Parker et al. 1996; Wang et al. 2000). A familial inheritance has been reported in 6–23% of patients with keratoconus, which reveals a genetic heterogeneity from autosomal dominant, with incomplete penetrance to autosomal recessive, but most reported keratoconus cases are sporadic (Bawazeer et al. 2000; Rabinowitz 1998; Wang et al. 2000; Wilson et al. 1992).
Six keratoconus susceptibility genomic loci, 20p11.21 (KTCN 1, MIM 148300), 16q22.3-q23.1 (KTCN 2, MIM 608932), 3p14-q13 (KTCN 3, MIM 608586), 2p24 (KTCN 4, MIM 609271), 5q14.3-q21.1, and 15q22.23-q24, have been previously identified through linkage studies of multigenerational families with keratoconus (Brancati et al. 2004; Dash et al. 2006; Heon et al. 2002; Hughes et al. 2003; Hutchings et al. 2005; Tang et al. 2005; Tyynismaa et al. 2002). Based on studies reporting significant linkage of keratoconus, several pathogenic mutations in VSX1 (MIM 605020; visual system homeobox gene 1, zebrafish, homolog of), located on 20p11.21, have been detected in multigenerational pedigrees of keratoconus, with a strong association and genetic variants of VSX1 that has been validated by multiple studies (Aldave et al. 2006; Bawazeer et al. 2000; Bisceglia et al. 2005; Chwa et al. 2006; Heon et al. 2002; Kenney and Brown 2003; Kuming and Joffe 1977; Liskova et al. 2007; Tang et al. 2008). Although several variants of VSX1, e.g., L17P, L159 M, and R166 W, have been reported to be pathogenic in multigenerational pedigrees of keratoconus with various associations, the identified pathogenic mutations has shown various associations in familial keratoconus patients with different ethnicities (Aldave et al. 2006; Bisceglia et al. 2005; Heon et al. 2002; Liskova et al. 2007; Tang et al. 2008). The association between variations in the VSX1 gene and keratoconus in Korean patients, however, has yet to be investigated. Therefore, it is of importance to identify the genetic factors that determine the susceptibility to keratoconus in Korean patients to gain insight into the pathogenesis of keratoconus. In particular, VSX1 is expressed in vitro and in vivo during corneal wound healing and in the corneal stroma, which may shed light on the pathophysiological origin of keratoconus (Barbaco et al. 2006). In this study, we assessed the influence of VSX1 variants on the incidence of sporadic keratoconus in Korean patients by screening the VSX1 gene.
Materials and methods
Subjects
This study consisted of 249 unrelated keratoconus patients and 208 controls who were all of Korean ancestry. All keratoconus patients, and controls lacking corneal disease, were identified from the Korea Eye Tissue and Gene Bank Related to Blindness, Department of Ophthalmology, the Catholic University of Korea. Appropriate informed consent was obtained from each subject, and all studies were performed according to the tenets of the Declaration of Helsinki. The patients were diagnosed with keratoconus based on the following criteria: (1) symptoms of keratoconus (Munson sign, protrusion, Vogt’s striae, corneal thickness, scarring, Fleishcher ring, photokeratoscopy signs, video keratography signs, and refractive errors) and (2) medical histories (age, gender, contact lense wearing, eye rubbing, systemic disease, atopy, and connective tissue disease (McMahon et al. 2006; Rabinowitz 1998). Control individuals were selected from the general population without keratoconus.
Mutation screening of the VSX1 gene
Genomic DNA was extracted from peripheral blood samples using the QIAamp DNA blood kit (QIAGEN, Valencia, USA). All polymerase chain reaction (PCR) studies were carried out with 25 ng of genomic DNA as a template in a mixture of PCR buffer, 2.5 mM magnesium chloride (MgCl2), 200 nM deoxynucleotide triphosphate (dNTP), 0.4 pmol of each primer, and 0.75 U of h-Taq polymerase (Solgent, Daejeon, Korea). To search mutations in this gene, we designed seven sets of primers that were used to amplify the PCR products of the entire coding region as well as exon–intron junctions of the gene (Table 1) (Bisceglia et al. 2005). The genotypes of single nucleotide polymorphisms (SNPs) spanning the coding region of the VSX1 gene were determined by single-strand conformational polymorphism (SSCP) analysis and direct sequencing. For SSCP, the amplified products were mixed with loading buffer with the addition of formamide, denatured at 95°C for 5 min, cooled in ice, and separated on a nondenaturing polyacrylamide gel. After electrophoresis, DNA bands were visualized by the silver staining method, and then those showing variation were directly sequenced. For DNA sequencing, amplified DNA was purified using the QIAquick PCR purification kit (Qiagen, Valencia, USA) and sequenced directly according to the protocols accompanying the BigDye Terminator cycle sequencing kit (Applied Biosystems, Foster City, CA, USA). The novel mutation G160V was confirmed by analysis of the BslI restriction fragment length polymorphism (RFLP). Nucleotide sequences were compared with the published VSX1 sequences (GenBank accession number NM_014588).
Statistics
To determine statistically significant differences between the groups by genotyping of SNPs and haplotypes, we used the χ2 test and 2 × 2 and 2 × m Fisher’s exact test for the contingency table file. The 2 × 2 contingency tables for each individual allele and the 2 × m contingency tables for each locus were used, where m refers to the number of marker alleles detected in the population. The Hardy–Weinberg equilibrium (HWE) of SNPs in patients and controls was calculated using the GenePop program. Values of p < 0.05 were considered statistically significant. The strength of the association was estimated by odds ratio (OR) of risk and 95% confidence intervals (CI) (JavaStat, http://members.aol.com/johnp71/ctab2x2.html). Haplotype frequencies and linkage disequilibrium measures were estimated using the Haploview package (Barrett et al. 2005).
Results
The 249 unrelated keratoconus patients and 208 controls recruited for this study had their entire VSX1 gene coding region and the exon–intron junctions sequenced by PCR-direct sequencing. Screening in the coding region of the VSX1 gene identified two heterozygous nonsynonymous mutations in exon 2 N151S (EX2 + 28A > G) and G160V (EX2 + 55G > T), and three heterozygous synonymous substitution, L176L (EX3 + 25G > A), A182A (rs12480307, EX3 + 43G > A) in exon 3 and G239G (EX4 + 90G > A) in exon 4 (Fig. 1). Exons 1 and 5 did contained no mutations.

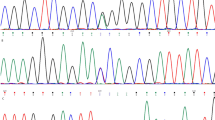
Electropherograms showing mutations by direct sequencing of the VSX1 gene exonic region: a N151S mutation, b G160 V mutation, c A182A polymorphism, d G239G polymorphism
The allele and genotype distribution and OR of these variants are given in Tables 2 and 3. In exon 2, the heterozygous transversion of EX2 + 55G > T (G160V) lies near the positively charged VSX1 nuclear localization signal and was present in 5.3% of unrelated keratoconus patients. Another nonsynonymous mutation, EX2 + 28A > G (N151S), was found in only one keratoconus patient (0.4%). Two nonsynonymous mutations, N151S and G160V, were not found in unaffected individuals from the control group.
Although the heterozygous mutations of EX3 + 25G > A (L176L) and EX3 + 43G > A (A182A) in the homeobox domain and EX4 + 90G > A (G239G) in the CVC domain were identified in affected individuals, these mutations were polymorphisms, the frequency of which did not differ significantly between cases and controls (Table 2).
In this study, we also found four intronic nucleotide changes, IVS1-11 t > a, IVS3 + 22 c > t, IVS3-23 g > a (rs6138482), and IVS3 + 84 t > a (rs62217198) (Fig. 2, Table 3). The IVS1-11*t/*a genotype was significantly higher in keratoconus patients than in the control group (7.2 vs. 1.0%, p = 0.001, OR = 8.03, 95% CI: 2.042 ≪ 31.432), and the IVS1-11*a allele frequency was higher in keratoconus patients compared with controls (3.6 vs. 0.5%, p = 0.001, OR = 7.76 95% CI: 1.989–30.241). There were no significant genotype or allele frequency differences at IVS3 + 22 c > t, IVS3 + 23 g > a, and IVS3 + 84 t > a between keratoconus patients and control subjects. The genotype distributions of all variants of VSX1 among the control subjects and the affected individuals were in Hardy–Weinberg equilibrium.

Electropherograms showing mutations by direct sequencing of the VSX1 gene intronic region: a IVS1-11 t > a polymorphism, b IVS3 + 22 c > t and IVS3 + 23 g > a polymorphisms, c IVS3 + 84 t > a polymorphism
In haplotype analysis, we identified two haplotype block sets: IVS1-11t > a, N151S (EX2 + 28A > G), G160V (EX2 + 55G > T), L176L (EX3 + 25G > A) for block A and A182A (EX3 + 43G > A), IVS3 + 22c > t, IVS3 + 23 g > a, IVS3 + 84t > a for block B. The AAGG of block A was more prevalent in keratoconus patients than in control subjects and with higher risk to keratoconus (p = 0.004, OR = 5.85, 95% CI 1.475 ≪ 23.151). The TATG of block A was present in only keratoconus patients (p < 0.001), whereas AAGG (p = 0.001) of block A and ACGT (p = 0.021) of block B were less frequent in the keratoconus compared with the control individuals (Table 4).
Discussion
The VSX1 gene, also named the retinal inner nuclear layer homeobox gene, is part of a paired-like subfamily of homeodomain transcription factors and contains a CVC domain (Hayashi et al. 2000; Passini et al. 1998; Semina et al. 2000). VSX1 is expressed in the embryonic craniofacial area and is restricted to cone bipolar cells of the inner nuclear layer, where it maintains function in the adult retina (Hayashi et al. 2005; Mintz-Hittner et al. 2004). Recently, Hosseini et al. (2008) reported the assessment of VSX1 expression in neonatal human cornea during perinatal endothelial differentiation. Barbaro et al. (2006) suggested that VSX1 is expressed in vitro and in vivo during human corneal wound healing, a process in which differentiation of corneal keratocytes into myofibroblasts occurs, but not in freshly obtained normal corneas. These results suggest a strong correlation between VSX1 expressions and wound-healing responses.
The VSX1 gene located within 20p11.21 of the candidate gene region for keratoconus and several pathogenic mutations in VSX1 gene were also found in various corneal dystrophies such as keratoconus and posterior polymorphous corneal dystrophy (PPCD, MIM122000) (Aldave 2005; Aldave et al. 2005; Heon et al. 2002). Heon et al. (2002) identified mutations in the VSX1 homeobox gene in patients with either keratoconus or PPCD. The R166W mutation responsible for keratoconus altered the homeodomain and impaired DNA binding. This decreased DNA binding was demonstrated by an electrophoretic mobility shift assay. Two mutations, L159M and G160D, near the nuclear localization signal of the VSX1 gene were associated with keratoconus and PPCD, respectively. Bisceglia et al. (2005) evaluated the role of the VSX1 gene in a series of 80 keratoconus-affected Italian subjects. Three described missense mutations, D144E, G160D, and P247R, were identified, and the novel mutation L17P was found in 8.7% of unrelated patients and three families. This mutation is transmitted as an autosomal dominant trait with variable expressivity and incomplete penetrance. Although Aldave et al. (2006) identified only one mutation, D144E, in a single affected keratoconus patient, D144E is a non-disease-causing polymorphism. Liskova et al. (2007) reported the D144E mutation and two novel intronic SNPs in a white British family, but these variants did not cosegregate with the disease phenotype. Tang et al. (2008) could not confirm the association of the three polymorphisms L159 M, R166 W, and H244R in the VSX1 gene in a larger pedigree with keratoconus because the distribution of the three polymorphisms was not significant enough to support a pathogenetic role in keratoconus. Aldave et al. (2006) and Tang et al. (2008) reported that the previously identified pathogenic mutations did not observe the association with sporadic keratoconus patients. Therefore, it is important to identify the genetic factors that determine susceptibility to keratoconus to gain insight into the pathogenesis of keratoconus in Korean patients because the susceptibility of mutations in VSX1 can vary in different ethnicities.
In this study, we report the results of a mutation screening of the VSX1 gene in unrelated Korean patients with keratoconus. Mutational analysis of the VSX1 gene revealed six novel mutations, EX2 + 28A > G (N151S), EX2 + 55G > T (G160V), EX3 + 25G > A (L176L), EX4 + 90G > A (G239G), IVS1-11t > a, and IVS3 + 22c > t, and three previously described changes, EX3 + 43G > A (A182A, rs12480307), IVS3 + 23G > A (rs6138482), and IVS3 + 84 t > a (rs622177198). Among these mutations, the heterozygous two-base-pair transition of EX2 + 55G > T near the positively charged VSX1 nuclear localization signal of exon 2 results in the substitution of glycine by valine (G160V). G160V was present in 5.3% of unrelated keratoconus patients, whereas it was not found in unaffected individuals from the control group or from any patients with other corneal diseases. The N151S mutation was present in 0.4% of unrelated keratoconus patients. In intragenic variants, IVS1-11*t/*a genotype (p = 0.001, OR = 8.03, 95% CI: 2.042 ≪ 31.432) and IVS1-11*a allele (p = 0.001, OR = 7.76, 95% CI: 1.989–30.241) frequencies were significantly higher in keratoconus patients than in the control group. With the haplotype analysis, the risk haplotypes, carring IVS1-11*a and EX2 + 55*T, were identified in our Korean study population. The AAGG and TATG haplotypes of IVS1-11t > a, EX2 + 28A > G (N151S), EX2 + 55G > T (G160V), and EX3 + 25G > A (L176L) combination were significantly associated with keratoconus. However, the common TAGG haplotype was found to be significantly decreased in keratoconus. These results suggest a role of VSX1 variants in the risk of unrelated Korean patients developing keratoconus and identify IVS1-11t > a and G160V variants as putative susceptibility variants. Both individual genotype data and corresponding haplotype data are consistent with a contribution of the IVS1-11t > a and G160V variants to keratoconus risk. Although in this study none of the mutations previously described as being mutations associated with keratoconus were identified in any of the affected patients, we found new mutations of the VSX1 gene in unrelated Korean keratoconus patients were considerably different from other ethnic groups.
Our data is the first report of VSX1 mutation screening in Korean keratoconus patients and detected novel missense mutation G160V and intragenic polymorphism IVS1-11t > a in the VSX1 gene. These show strong statistical association with unrelated keratoconus patients. Consequently, the study suggests that two novel variants, IVS1-11t > a and G160V, in the VSX1 gene seem to be possible candidate variants for keratoconus predisposition in unrelated Korean patients. However, it is not known how SNPs in the VSX1 gene might control progression and onset of keratoconus, and they clearly require further analysis to elucidate their relevance to this impairment.
References
Aldave AJ (2005) VSX1 mutation and corneal dystrophies. Ophtahlmology 112:170–171
Aldave AJ, Yellore VS, Principe AH, Abedi G, Merrill K, Cjalukya M, Small KW, Udar N (2005) Candidate gene screening for posterior polymorphous dystrophy. Cornea 24:151–155
Aldave AJ, Yellore VS, Salem AK, Yoo GL, Rayner SA, Yang H, Tang GY, Piconell Y, Raninowitz YS (2006) No VSX1 gene mutations associated with keratoconus. Invest Ophthalmol Vis Sci 47:2820–2822
Barbaro V, Di Iorio E, Ferrari S, Bisceglia L, Ruzza A, De Luca M, Pellegrini G (2006) Expression of VSX1 in human corneal keratocytes during differentiation into myofibroblasts in response to wound healing. Invest Ophthalmol Vis Sci 47:5243–5250
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Bawazeer AM, Hodge WG, Lorimer B (2000) Atopy and keratoconus: a multivariate analysis. Br J Ophthalmol 84:834–836
Bisceglia L, Ciaschetti M, De Bonis P, Campo PA, Pizzicoli C, Scala C, Grifa M, Ciavarella P, Delle Noci N, Vaira F, Macaluso C, Zelante L (2005) VSX1 mutational analysis in a series of Italian patients affected by keratoconus: detection of a novel mutation. Invest Ophthalmol Vis Sci 46:39–45
Brancati F, Valente EM, Sarkozy A, Feher J, Castory M, Del Duca P, Mingarelli R, Pizzuti A, Dallapiccola B (2004) A locus for autosomal dominant keratoconus maps to human chromosome 3p14–q13. J Med Genet 41:188–192
Chwa M, Atilano SR, Reddy V, Jordan N, Kim DW, Kenney MC (2006) Increased stress-induced generation of reactive oxygen species and apoptosis in human keratoconus fibroblasts. Invest Ophthalmol Vis Sci 47:1902–1910
Dash DP, Silvestri G, Hughes AE (2006) Fine mapping of the keratoconus with cataract locus on chromosome 15q and candidate gene analysis. Mol Vis 12:499–505
Edwards M, McGhee CN, Deen S (2001) The genetics of keratoconus. Clin Exp Ophthlamol 29:345–351
Elder MJ (1994) Leber congenital amaurosis and its association with keratoconus and keratoglobus. J Pediatr Ophthalmol Strabismus 31:38–40
Flanders M, Lapointe ML, Brownstein S, Little JM (1084) Keratoconus and Leber’s congenital amaurosis: a clinicopathological correlation. Can J Ophthalmol 19:310–314
Hayashi T, Huang J, Deeb SS (2000) RINX (VSX1), a novel homeobox gene expressed in the inner nuclear layer of the adult retina. Genomics 67:128–139
Hayashi T, Huang J, Deeb SS (2005) Expression of Rinx/Vsx1 during postnatal eye development in cone-bipolar, differentiating ganglion, and lens fiber cells. Jpn J Ophthalmol 49:93–105
Heon E, Greenberg A, Kopp KK, Rootman D, Vincent AL, Billingsley G, Priston M, Dorval KM, Chow RL, McInnes RR, Heathcote G, Westall C, Sutphin JE, Semina E, Bremner R, Stone EM (2002) VSX1, a gene for posterior polymorphous dystrophy and keratoconus. Hum Mol Genet 11:1029–1036
Hosseini SM, Herd S, Vincent AL, Heon E (2008) Genetic analysis of chromosome 20-related posterior polymorphous corneal dystrophy: genetic heterogeneity and exclusion of three candidate genes. Mol Vis 14:71–80
Hughes AE, Dash DP, Jackson AJ, Frazer DG, Silvestri G (2003) Familial keratoconus with cataract: linkage to the long arm of chromosome 15 and exclusion of candidate genes. Invest Ophthalmol Vis Sci 44:5063–5066
Hutchings H, Ginisty H, Le Gallo M, Levy D, Stoesser F, Rounland JF, Arne JL, Lalaux MH, Calvas P, Roth MP, Hovnanian A, Malecaze F (2005) Identification of a new locus for isolated familial keratoconus at 2p24. J Med Genet 42:88–94
Karseras AG, Ruben M (1976) Aetiology of keratoconus. Br J Ophthalmol 60:522–525
Kenney MC, Brown DJ (2003) The cascade hypothesis of keratoconus. Contact Lens Anterior Eye 26:139–146
Krachmer JH, Feder RS, Belin MW (1984) Keratoconus and related noninflammatory corneal thinning disorders. Surv Ophthalmol 28:293–322
Kuming BS, Joffe L (1977) Ehlers-Danlos syndrome associated with keratoconus. A case report. S Afr Med J 52:403–405
Liskova P, Ebenezer ND, Hysi PG, Gwilliam R, El-Ashry MF, Moodaley LC, Hau S, Twa M, Tuft SJ, Bhatacharya SS (2007) Molecular analysis of the VSX1 gene in familial keratoconus. Mol Vis 13:1887–1891
McMahon TT, Shin JA, Newlin A, Edrington TB, Sugar J, Zadnik K (1999) Discordance for keratoconus in two pairs of monozygotic twins. Cornea 18:444–451
McMahon TT, Szczotka-Flynn L, Barr JT, Anderson RJ, Slaughter ME, Lass JH, Iyengar SK et al (2006) A new method for grading the severity of keratoconus: the Keratoconus Severity Score (KSS). Cornea 25:794–800
Mintz-Hittner HA, Semina EV, Frishman LJ, Prager TC, Murray JC (2004) VSX1 (RINX) mutation with craniofacial anomalies, empty sella, corneal endothelial changes, an abnormal retinal and auditory bipolar cells. Ophthalmology 111:828–836
Nakamura H, Riley F, Sakai H (2005) Histopathological and immunohistochemical studies of lenticules after epikeratoplasty for keratoconus. Br J Ophthalmol 89:841–846
Parker J, Ko W, Pavlopoulos G, Wolfe PJ, Rabinowitz YS, Feldman ST (1996) Videokeratography of keratoconus in monozygotic twins. J Refract Surg 12:180–183
Passini MA, Kurtzman AL, Canger AK, Asch WS, Wray GA, Raymond PA, Schechter N (1998) Cloning of zebrafish vsx1: expression of a paired-like homeobox gene during CNS development. Dev Genet 23:128–141
Rabinowitz YS (1998) Keratoconus. Surv Ophthalmol 42:297–319
Semina EV, Mintz-Hittner HA, Murray JC (2000) Isolation and characterization of a novel human paired-like homeodomain-containing transcription factor gene, VSX1, expressed in ocular tissues. Genomics 63:289–293
Shapiro MB, France TD (1985) The ocular features of Down’s syndrome. Am J Ophthalmol 99:659–663
Sherwin T, Brookes NH (2004) Morphological changes in keratoconus: pathology or pathogenesis. Clin Experiment Ophthalmol 32:211–217
Tang YG, Picornell Y, Su X, Li X, Yang H, Rabinowitz YS (2008) Three VSX1 gene mutations, L159M, R166W, and H244R, are not associated with keratoconus. Cornea 27:189–192
Tang YG, Rabinowitz YS, Taylor KD, Li X, Hu M, Picornell Y, Yang H (2005) Genomewide linkage scan in a multigeneration Caucasian pedigree identifies a novel locus for keratoconus on chromosome 5q14.3–q21.1. Genet Med 7:397–405
Tyynismaa H, Sistonen P, Tuupanen S, Tervo T, Dammert A, Latvala T, Alitalo T (2002) A locus for autosomal dominant keratoconus: linkage to 16q22.3–23.1 in Finnish families. Invest Ophthalmol Vis Sci 43:3160–3164
Wang Y, Rabompwitz YS, Rotter JI, Yang H (2000) Genetic epidemiological study of keratoconus: evidence for major gene determination. Am J Med Genet 93:403–409
Wilson SE, Verity SM, Conger DL (1992) Accuracy and reproducibility of the corneal analysis system and topographic modeling system. Cornea 11:28–35
Acknowledgments
We thank the patients who participated in the study. This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD) (KRF-2006-E00016). We thank the Korea Eye Tissue and Gene Bank, The Catholic University of Korea, and the staff of the Department of Ophthalmology, St. Mary’s Hospital.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mok, JW., Baek, SJ. & Joo, CK. VSX1 gene variants are associated with keratoconus in unrelated Korean patients. J Hum Genet 53, 842–849 (2008). https://doi.org/10.1007/s10038-008-0319-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-008-0319-6
Keywords
This article is cited by
-
Do age-related macular degeneration genes show association with keratoconus?
Eye and Vision (2019)
-
Genetic Aspects of Keratoconus: A Literature Review Exploring Potential Genetic Contributions and Possible Genetic Relationships with Comorbidities
Ophthalmology and Therapy (2018)
-
Analysis of the VSX1 gene in sporadic keratoconus patients from China
BMC Ophthalmology (2017)
-
Genetic associations for keratoconus: a systematic review and meta-analysis
Scientific Reports (2017)
-
Genomic strategies to understand causes of keratoconus
Molecular Genetics and Genomics (2017)
