
Abstract
Deletions of the SHOX gene (Xp22-Yp11.3) are associated with Leri–Weill dyschondrosteosys (LWD) and idiopathic short stature. It has been estimated that SHOX deletions occur in 1,000–2,000 individuals in the total population, suggesting that this alteration should be investigated in all cases with unexplained short stature. SHOX deletions are currently investigated using fluorescence in situ hybridization (FISH) or molecular analysis of intragenic CA repeats. However, both techniques show some limitations. In the present study, the use of the multiple ligation probe amplification (MLPA) assay for the identification and characterization of SHOX deletions in 15 LWD patients, 3 of which carriers of chromosome abnormalities involving the SHOX gene, is reported. MLPA analysis demonstrated the heterozygous deletion of SHOX in seven patients (46.6%), disclosing the presence of two different proximal breakpoints. In patients with abnormal karyotype, MLPA analysis was able to identify the chromosomal rearrangement, showing, in addition to the SHOX deletions, the gain or loss of other genes mapped on the X and Y chromosomes. Since MLPA analysis can be carried out on a simple buccal swab, avoiding invasive peripheral blood collection, this technique represents a fast, simple and high throughput approach in the screening of SHOX deletions, able to provide more information as compared to FISH and microsatellite analysis.
Similar content being viewed by others


Introduction
The human short stature homeoboX-containing gene (SHOX ) is located in the pseudoautosomal region (PAR1) of sex chromosomes at Xp22.3-Yp11.3 (Rao et al. 1997; Ellison et al. 1997). SHOX is composed of 6 exons spanning 35 Kb, and produces 2 alternatively spliced mRNA forms encoding for 2 transcription factors (SHOXa and SHOXb) of 292 and 225 amino acids, respectively (Rao et al. 1997, 2001).
Haploinsufficiency of the SHOX gene was originally associated with short stature in Turner syndrome and in some cases of idiopathic growth retardation (Rao et al. 1997; Ellison et al. 1997). Subsequently, SHOX mutations were correlated with mesomelic short stature and Madelung deformity in the Leri–Weill dyschondrosteosys (LWD) (OMIM #127300) (Belin et al. 1998; Shears et al. 1998). Different reports have demonstrated that SHOX mutations are detectable in 50–90% of patients with LWD (Blaschke and Rappold 2006; Ross et al. 2001; Cormier-Daire et al. 2001; Falcinelli et al. 2002; Flanagan 2002) and in 2–12% of patients with idiopathic short stature (Rappold et al. 2002; Stuppia et al. 2003; Morizio et al. 2003; Binder et al. 2003). About 80% of SHOX mutations are complete gene deletions, whereas diverse point mutations account for the remaining cases (Ross 2001; Stuppia 2003).
In a proportion of cases, SHOX deletions derive from chromosomal rearrangements involving the PAR1 (Calabrese et al. 1999; Palka et al. 2000; Rehiner et al. 2001; Shago et al. 2002; Doherty et al. 2003; Borie et al. 2004; Kokalj-Vokac ey al. 2004; Fukami et al. 2005). The characterization of these rearrangements is of crucial importance, since the involvement in the rearrangement of other genes mapped on sex chromosomes can produce additional clinical features, mainly resembling Turner syndrome (Palka et al. 2000).
SHOX deletions are usually detected by fluorescence in situ hybridization (FISH) or molecular analysis of intragenic CA repeats. However, neither technique is able to provide a clearcut identification of the deletion breakpoints nor to disclose the involvement of other genes in the rearrangement.
In recent years, the multiple ligation probe amplification (MLPA) technique (Schouten et al. 2002; Sellner and Taylor 2004) has been demonstrated as a useful tool in the detection and characterization of deletions or duplications of several genes, such as BRCA1 (Hogervorst et al. 2003), DMD (Gatta et al. 2005), PMP22 (Slater et al. 2004), and others. This approach, based on the simultaneous hybridization of different probes followed by a multiplex PCR amplification, allows the establishment of the copy number of the target gene(s) and of several control genes.
Here, we report on the use of the MLPA assay in 15 LWD patients for the detection of SHOX deletions, the identification of break points, and the characterization of the rearrangements.
Materials and methods
Patients
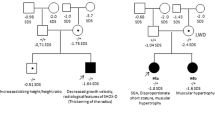
Fifteen patients, 11 females and 4 males, averaging 13 years (range 7–34 years) entered this study (patients 1–15). Clinical, cytogenetics and molecular features of nine of these patients had been previously reported (Calabrese et al. 1999; Palka et al. 2000; Stuppia et al. 2003; Morizio et al. 2003). All patients had typical LWD, diagnosed on the basis of previously reported criteria (Palka et al. 2000), except patient 3, who also showed features of Turner Syndrome. Twelve patients had normal karyotype (patients 4–15) and three (patients 1–3) had an abnormal karyotype consisting, in patients 1–2, a mother and her son, of a t(X; Y)(p22.3; q11), and in patient 3 of a 46, X, i(Xq). These latter cases with abnormal karyotype were included in the study in order to verify the ability of the MLPA approach to also detect, in addition to the SHOX gene loss, the presence of other alterations, and to correctly identify the chromosome rearrangement.
FISH, with the 34F5 SHOX specific probe (gift of Dr. Gudrun Rappold), had been performed in 9 out of the 15 patients, 7 with normal karyotype (patients 4–5, 8–12) and 2 with the t(X; Y)(p22.3; q11). Deletion of one copy of SHOX was detected in 4 cases (patients 1–2, 4–5).
MLPA analysis
DNA of the 15 LWD patients and of 10 healthy controls (5 males and 5 females) was obtained from peripheral blood cells using standard procedures, or from buccal swabs using the MasterAmp Buccal Swab kit (Epicentre, Madison, USA). MLPA analysis was carried out using the SALSA P018 SHOX MLPA Kit (MRC Holland, Amsterdam, The Netherlands), containing 13 probes specific for the PAR 1, and 29 control probes, which are simultaneously used in a single reaction. Two of the PAR1 specific probes (PR48 and LOC159015) are telomeric to SHOX (probes 1–2), eight are specific for SHOX gene (probes 3–10), and three (CSF2RA, IL3RA and ASMT) are proximal to SHOX (probes 11–13). Twelve out of the 29 control probes are specific for sex chromosomes, namely 3 probes for OA1 gene (Xp22.3), 1 probe each for genes PPEF1 (Xp22), AR (Xq11-q12), PDCD8 (Xq25), L1CAM (Xq28), SYBL1 (PAR 2), SRY (Yp11.3), UTY (Yq11) and SMCY (Yq11), and 1 further control probe for the X chromosome. The remaining 17 probes are specific for autosomal loci. MLPA reaction were carried out according to the manufacturer's recommendations. All samples were run using a 310 ABI PRISM Genetic Analyzer and analyzed by the GeneScan 2.1 Software (Applied Biosystems). For each sample, relative peaks area was calculated and compared to five sex-matched controls using the Coffalyser 1.4 software (MRC Holland). This program identifies a peak as normal when showing a 0.7–1.3 ratio with normal controls, deleted when showing a ratio <0.7 and duplicated when showing a ratio >1.3. In male patients, hemizygous deletions of X- and Y-specific sequences result in the absence of the relative peaks.
Results
Multiple ligation probe amplification analysis demonstrated the heterozygous deletion of the SHOX gene in 7 out of the 15 (46.6%) LWD patients (patients 1–7), showing all the peaks specific for SHOX exons at a ratio <0.7 as compared to normal controls. Based on the results shown by the other PAR1 specific probes, two different size deletions were detected. In fact, five cases (patients 1–3, 6–7) showed a deletion encompassing the PAR1 probes 1–13, mapping the proximal breakpoint between ASMT and SRY genes in the male patients, and between ASMT and OA1 genes in female patients. In the remaining two cases (patients 4–5), MLPA showed the deletion of probes 1–10, with proximal breakpoint between SHOX and CSF2RA. Due to the lack of a specific p-tel probe in the kit, it was not possible to identify whether these rearrangements were terminal deletions or interstitial deletions with telomeric breakpoint between p-tel and PR48 (Fig. 1).

Deletion map of the seven LWD patients showing SHOX deletion. White bars indicate deleted regions. Black bars indicate retained regions. Grey bars indicate sequences whose involvement in the deletion cannot be established on the basis of MLPA results. The localization of MLPA probes on the X chromosome and the results showed by a female control and six females LWD patients are reported in the top of the figure. The localization of MLPA probes in the Y chromosome and the results showed by a male control and one male LWD patients are reported at the bottom of the figure. * patient carrier of a 46, X -X, + der(X),t(X;Y)(p22.3;q11); ° patient carrier of a 46, X, i(Xq); # patient carrier of a 46, Y, -X, + der(X),t(X; Y)(p22.3; q11)
In three cases (patients 1–3), all carriers of chromosomal abnormalities, MLPA analysis also showed, in addition to the deletion of the PAR1 sequences, the involvement of other X or Y chromosome regions. One female LWD patient (patient 1) showed a duplication of SYBL1 (PAR2) and the presence of the Y-specific SMCY gene, indicating the presence of three copies of the PAR2 and of material from Yq including SMCY gene. The son of this patient (patient 2) showed a duplication of SYBL1 and SMCY signals, with size peaks indicating the presence of a single copy of the other Y-specific sequences (SRY and UTY) and of all the X-specific sequences. These results agree with cytogenetic data showing a der (X) chromosome in both patients, with the second sex chromosome being an X in the mother and a Y in the son. MLPA analysis also allowed the mapping of the break on the Y chromosome between UTY and SMCY genes, the latter being present on the der (X). In patient 3, a carrier of an i(Xq), MLPA analysis showed the heterozygous deletion of the entire PAR1 and of all the Xp specific regions, with duplication of the PAR2 and of all Xq specific sequences. These results demonstrated the presence in the patients of a single copy of Xp, with three copies of Xq, typical of an i(Xq).
All the obtained results were identical in peripheral blood samples and in those obtained by buccal swab.
Detailed results of MLPA analysis are reported in Table 1 and Fig. 2.

Coffalyzer sheet showing the ratios calculated for each probe in all the investigated patiens (LWD 1–15) and in two normal controls (one male and one female)
Discussion
Deletions of the PAR1 encompassing SHOX gene have been mainly reported in patients with LWD or with idiopathic short stature. Based on the prevalence of SHOX deletions in patients with idiopathic short stature, it has been estimated that this alteration occurs in 1,000–2,000 individuals in the total population (Rappold et al. 2002; Binder et al. 2003). Due to this high prevalence, it has been suggested that SHOX deletions should be investigated in all children with unexplained short stature, since patients with SHOX haploinsufficiency could benefit from early GH treatment (Binder et al. 2000). However, the approach to be used for such a screening is still under debate. Cytogenetic analysis is useful for the identification of large chromosomal rearrangements involving the PAR1 (Palka et al. 2000), but cannot detect small intragenic deletions. FISH analysis is also able to identify SHOX deletions on interphase nuclei from a blood-smear (Morizio et al. 2003), but is a low throughput technique requiring a specific probe not commercially available. Finally, microsatellite analysis requires DNA from the proband’s parents and cannot disclose large chromosomal rearrangements underlying SHOX deletion. Very recently, a quantitative Real-Time PCR assay for the detection of SHOX haploinsufficiency has been described, confirming the necessity of alternative approaches for the study of this gene (Tan and Loke 2005).
In the present study, we investigated the usefulness of the MLPA assay for the identification and characterization of SHOX deletions in LWD patients both those with normal karyotype and carriers of chromosomal rearrangements. In patients with normal karyotype, MLPA was able to confirm all SHOX deletions previously detected by FISH and to also disclose deletions in two patients not previously analyzed. In addition, this approach provided more information about the size and the breakpoints of the deletions. In fact, in our series, MLPA analysis showed two different proximal breakpoints, the first one mapped between SHOX and CSF2RA, and the second one between ASMT and OA1 in females and between ASMT1 and SRY in males. Both deletions caused the loss of the entire SHOX gene, and in no case were partial deletions detected, suggesting that intragenic SHOX deletions are not a common cause of LWD.
A previous study investigating the breakpoints localization in SHOX deletions mapped, in 73% of cases, the proximal breakpoint less than 100 kb to SHOX (Schneider et al. 2005). This hotspot is located between SHOX and CSF2RA, where MLPA analysis mapped the proximal breakpoint in two patients of our series. Larger deletions, with proximal breakpoints mapped more than 2 Mb to SHOX, were detected by Schneider et al. (2005) in a minority of the investigated cases. These proximal breakpoints correspond to those mapped between ASMT and OA1 in the other patients of our series. In a very recent report, Zinn et al. (2006) described an additional hotspot for PAR1 deletion about 1,200 kb to SHOX. Due to the lack of probes mapped between SHOX and CSF2RA in the MLPA kit, it is not possible to precisely identify this breakpoint in our series. However, the two deletions removing probes 1–10 could have their proximal breakpoint located in the region indicated by Zinn et al. (2006). Thus, MLPA results are in agreement with literature data concerning the presence of different proximal breakpoints in SHOX deletions, although the specific localization of these breakpoints has been recently discussed (Benito-Sanz et al. 2006). As to the distal breakpoint, the absence of a specific telomeric probe within the MLPA kit did not allow the discrimination of terminal from interstitial deletions in our series.
Multiple ligation probe amplification analysis was very informative in the study of three LWD patients’ carriers of chromosomal rearrangements. In fact, in two patients with a t(X,Y), MLPA was able to identify the chromosomal rearrangement and to refine the deletion breakpoints on both the X and Y chromosomes. Also, in the analysis of the patient with the i(Xq), MLPA confirmed the cytogenetic result, showing the presence of a single copy of all Xp specific sequences and three copies of all Xq specific sequences. These results demonstrate that MLPA analysis is not only able to detect SHOX deletions, but also to disclose the presence of large chromosomal rearrangements involving sex chromosomes and to correctly identify the breakpoints.
In conclusion, MLPA analysis represents a simple, rapid and sensible tool for the detection of SHOX deletions. This analysis could be used for large scale screening of SHOX deletions since it can be performed on a simple buccal swab, avoiding invasive peripheral blood collection. Compared to FISH, MLPA is a high throughput analysis, allowing the simultaneous study of several samples in the same experiment and the investigation of different genes in each sample. Compared to microsatellite analysis, MLPA can also be carried out without DNA samples of the probands’ parents, and, furthermore, provides information about the deletion breakpoints and the presence of chromosomal rearrangements involving other X and Y chromosome specific sequences, which can produce clinical features additional to the typical LWD phenotype.
References
Belin V, Cusin V, Viot G, Girlich D, Toutain A, Moncla A, Vekemans M, Le Merrer M, Munnich A, Cormier-Daire V (1998) SHOX mutations in dyschondrosteosis (Leri–Weill syndrome). Nat Genet 19:67–69
Benito-Sanz S, Gorbenko del Blanco D, Huber C, Thomas NS, Aza-Carmona M, Bunyan D, Maloney V, Argente J, Cormier-Daire V, Campos-Barros A, Heath KE (2006) Characterization of SHOX deletions in Leri–Weill Dyschondrosteosis (LWD) reveals genetic heterogeneity and no recombination hotspots. Am J Hum Genet 79:409–414
Binder G, Schwarze CP, Ranke MB (2000) Identification of short stature caused by SHOX defects and therapeutic effect of recombinant human growth hormone. J Clin Endocrinol Metab 85:245–249
Binder G, Ranke MB, Martin DD (2003) Auxology is a valuable instrument for the clinical diagnosis of SHOX haploinsufficiency in school-age children with unexplained short stature. J Clin Endocrinol Metab 88:4891–4896
Blaschke RJ, Rappold G (2006) The pseudoautosomal regions, SHOX and disease. Curr Opin Genet Dev 16:233–239
Borie C, Leger J, Dupuy O, Hassan M, Ledu N, Lebbar A, Czernichow P, Eydoux P (2004) Translocation (Y; 22) resulting in the loss of SHOX and isolated short stature. Am J Med Genet A 125:186–190
Calabrese G, Fischetto R, Stuppia L, Capodiferro F, Mingarelli R, Causio F, Rocchi M, Rappold GA, Palka G (1999) X/Y translocation in a family with Leri–Weill dyschondrosteosis. Hum Genet 105:367–368
Cormier-Daire V, Huber C, Munnich A (2001) Allelic and nonallelic heterogeneity in dyschondrosteosis (Leri–Weill syndrome). Am J Med Genet 106:272–274
Doherty MJ, Glass IA, Bennett CL, Cotter PD, Watson NF, Mitchell AL, Bird TD, Farrell DF (2003) An Xp; Yq translocation causing a novel contiguous gene syndrome in brothers with generalized epilepsy, ichthyosis, and attention deficits. Epilepsia 44:1529–1535
Ellison JW, Wardak Z, Young MF, Gehron Robey P, Laig-Webster M, Chiong W (1997) PHOG, a candidate gene for involvement in the short stature of Turner syndrome. Hum Mol Genet 6:1341–1347
Falcinelli C, Iughetti L, Percesepe A, Calabrese G, Chiarelli F, Cisternino M, De Sanctis L, Pucarelli I, Radetti G, Wasniewska M, Weber G, Stuppia L, Bernasconi S, Forabosco A (2002) SHOX point mutations and deletions in Leri–Weill dyschondrosteosis. J Med Genet 39:E33
Flanagan SF, Munns CF, Hayes M, Williams B, Berry M, Vickers D, Rao E, Rappold GA, Batch JA, Hyland VJ, Glass IA (2002) Prevalence of mutations in the short stature homeobox containing gene (SHOX) in Madelung deformity of childhood. J Med Genet 39:758–763
Fukami M, Okuyama T, Yamamori S, Nishimura G, Ogata T (2005) Microdeletion in the SHOX 3′ region associated with skeletal phenotypes of Langer mesomelic dysplasia in a 45, X/46, X, r(X) infant and Leri–Weill dyschondrosteosis in her 46, XX mother: implication for the SHOX enhancer. Am J Med Genet A 137:72–76
Gatta V, Scarciolla O, Gaspari AR, Palka C, De Angelis MV, Di Muzio A, Guanciali-Franchi P, Calabrese G, Uncini A, Stuppia L (2005) Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA). Hum Genet 117:92–98
Hogervorst FB, Nederlof PM, Gille JJ, McElgunn CJ, Grippeling M, Pruntel R, Regnerus R, van Welsem T, van Spaendonk R, Menko FH, Kluijt I, Dommering C, Verhoef S, Schouten JP, van’t Veer LJ, Pals G (2003) Large genomic deletions and duplications in the BRCA1 gene identified by a novel quantitative method. Cancer Res 63:1449–1453
Kokalj-Vokac N, Marcun-Varda N, Zagorac A, Erjavec-Skerget A, Zagradisnik B, Todorovic M, Gregoric A (2004) Subterminal deletion/duplication event in an affected male due to maternal X chromosome pericentric inversion. Eur J Pediatr 163:658–663
Morizio E, Stuppia L, Gatta V, Fantasia D, Guanciali Franchi P, Rinaldi MM, Scarano G, Concolino D, Giannotti A, Verrotti A, Chiarelli F, Calabrese G, Palka G (2003) Deletion of the SHOX gene in patients with short stature of unknown cause. Am J Med Genet A 119:293–296
Palka G, Stuppia L, Guanciali Franchi P, Chiarelli F, Fischetto R, Borrelli P, Giannotti A, Fioretti G, Rinaldi MM, Mingarelli R, Rappold GA, Calabrese G (2000) Short arm rearrangements of sex chromosomes with haploinsufficiency of the SHOX gene are associated with Leri–Weill dyschondrosteosis. Clin Genet 57:449–453
Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A, Muroya K, Binder G, Kirsch S, Winkelmann M, Nordsiek G, Heinrich U, Breuning MH, Ranke MB, Rosenthal A, Ogata T, Rappold GA (1997) Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet 16:54–63
Rao E, Blaschke RJ, Marchini A, Niesler B, Burnett M, Rappold GA (2001) The Leri–Weill and Turner syndrome homeobox gene SHOX encodes a cell-type specific transcriptional activator. Hum Mol Genet 10:3083–3091
Rappold GA, Fukami M, Niesler B, Schiller S, Zumkeller W, Bettendorf M, Heinrich U, Vlachopapadoupoulou E, Reinehr T, Onigata K, Ogata T (2002) Deletions of the homeobox gene SHOX (short stature homeobox) are an important cause of growth failure in children with short stature. J Clin Endocrinol Metab 87:1402–1406
Reinehr T, Jauch A, Zoll B, Engel U, Bartels I, Andler W (2001) Short stature in a mother and daughter caused by familial der(X)t(X;X)(p22.1–3; q26). Am J Med Genet 102:81–85
Ross JL, Scott C Jr, Marttila P, Kowal K, Nass A, Papenhausen P, Abboudi J, Osterman L, Kushner H, Carter P, Ezaki M, Elder F, Wei F, Chen H, Zinn AR (2001) Phenotypes associated with SHOX deficiency. J Clin Endocrinol Metab 86:5674–5680
Schneider KU, Sabherwal N, Jantz K, Roth R, Muncke N, Blum WF, Cutler GB Jr, Rappold G (2005) Identification of a major recombination hotspot in patients with short stature and SHOX deficiency. Am J Hum Genet 77:89–96
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G (2002) Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 30:E57
Sellner LN, Taylor GR (2004) MLPA and MAPH: new techniques for detection of gene deletions. Hum Mutat 23:413–419
Shago M, Sgro M, Barozzino T, Antinucci D, Chakraborty P, Chitayat D, Teshima I (2002) An active ring X and haploinsufficiency of SHOX contribute to short stature, congenital anomalies, and developmental delay in a female. Am J Med Genet 113:279–285
Shears DJ, Vassal HJ, Goodman FR, Palmer RW, Reardon W, Superti-Furga A, Scambler PJ, Winter RM (1998) Mutation and deletion of the pseudoautosomal gene SHOX cause Leri–Weill dyschondrosteosis. Nat Genet 19:70–73
Slater H, Bruno D, Ren H, La P, Burgess T, Hills L, Nouri S, Schouten J, Choo KH (2004) Improved testing for CMT1A and HNPP using multiplex ligation-dependent probe amplification (MLPA) with rapid DNA preparations: comparison with the interphase FISH method. Hum Mutat 24:164–171
Stuppia L, Calabrese G, Gatta V, Pintor S, Morizio E, Fantasia D, Guanciali Franchi P, Rinaldi MM, Scarano G, Concolino D, Giannotti A, Petreschi F, Anzellotti MT, Pomilio M, Chiarelli F, Tumini S, Palka G (2003) SHOX mutations detected by FISH and direct sequencing in patients with short stature. J Med Genet 40:E11
Tan YM, Loke KY (2005) Quantitative real-time polymerase chain reaction (RQ-PCR) for the rapid detection of SHOX haploinsufficiency in Leri-Weill Syndrome. Diagn Mol Pathol 14:247–249
Zinn AR, Ramos P, Ross JL (2006) A second recombination hotspot associated with SHOX deletions. Am J Hum Genet 78:523–535
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gatta, V., Antonucci, I., Morizio, E. et al. Identification and characterization of different SHOX gene deletions in patients with Leri–Weill dyschondrosteosys by MLPA assay. J Hum Genet 52, 21–27 (2007). https://doi.org/10.1007/s10038-006-0074-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0074-5
Keywords
This article is cited by
-
Variants in the 5′UTR reduce SHOX expression and contribute to SHOX haploinsufficiency
European Journal of Human Genetics (2021)
-
Identification of a novel 15.5 kb SHOX deletion associated with marked intrafamilial phenotypic variability and analysis of its molecular origin
Journal of Genetics (2016)
-
Spectrum of phenotypic anomalies in four families with deletion of the SHOX enhancer region
BMC Medical Genetics (2014)
-
MS-MLPA analysis for FMR1 gene: evaluation in a routine diagnostic setting
BMC Medical Genetics (2013)
-
Identification of the second CFTR mutation in patients with congenital bilateral absence of vas deferens undergoing ART protocols
Asian Journal of Andrology (2010)
