
Abstract
Hereditary hemorrhagic telangiectasia (HHT) or Osler–Rendu–Weber disease is a systemic fibrovascular dysplasia with an autosomal dominant inheritance pattern. Mutations in two genes, endoglin and ALK-1, are known to cause HHT, both of which mediate signaling by transforming growth factor β ligands in vascular endothelial cells. Ten patients were analyzed. Diagnosis of HHT was carried out by means of family history, recurrent bleeding, and the presence of multiple telangiectases lesions. Conformation-sensitive gel electrophoresis analyses with consistent abnormal migration patterns were cloned and sequenced using the MegaBace 1000 DNA automated analyzer. Three novel mutations were identified in the coding sequence of the ALK-1 gene in five patients and their families, which demonstrated clinical manifestations of HHT type 2. These mutations included a G insertion and a T deletion of single base pairs in exons 3 and 7, as well as missense mutations in exons 7 and 8 of the ALK-1 gene. These data indicate that loss-of-function mutations in a single allele of the ALK1 locus are sufficient to contribute to defects in maintaining endothelial integrity. We suggest the high rate of mutation detection and the small size of the ALK-1 gene make genomic sequencing a viable diagnostic test for HHT2.
Similar content being viewed by others


Introduction
Hereditary hemorrhagic telangiectasia (HHT) or Osler–Rendu–Weber disease (ORW) is an autosomal dominant multisystemic vascular dysplasia (Plauchu et al. 1989; Guttmacher et al. 1995), characterized by severe recurrent nose bleeding, localized mucocutaneous telangiectases, gastrointestinal hemorrhage, as well as arteriovenous malformations (AVM) in the lungs, liver, gastrointestinal tract, and brain, which can cause severe ischemic injury or stroke (Guttmacher et al. 1995). Gastrointestinal bleeding occurs in approximately one third of patients, especially in later life, and is well recognized (Plauchu et al. 1989).
Estimates of the incidence of HHT vary widely, but studies have shown that it affects between 1 in 8,345 and 1 in 40,000 people with even a higher incidence in some isolated populations (Shovlin et al. 2000; Berg et al. 2003). The mortality rate as a direct result of the disease is about 36% (Sawabe et al. 2001). Furthermore, Kjeldsen et al. 1999 reported an increased mortality among young HHT patients when compared with those of patients older than 60 years.
Molecular heterogeneity of the disease has been revealed by linkage and mutational studies showing at least two distinct loci for HHT. The first locus, HHT-1, comprises the endoglin (ENG) gene mapped to chromosome 9q33 (McAllister et al. 1994a, b). Studies have shown that patients with HHT-1 are more likely to develop pulmonary arteriovenous malformations (PAVM) than patients with HHT-2 (Berg et al. 1996).
Endoglin is a TGF-β binding protein, expressed predominantly by endothelial cells and placenta (Gougos and Letarte 1990; Cheifetz et al. 1992). Loss-of-function mutations in the human ENG gene cause HHT type I, which is characterized by a higher prevalence of symptomatic PAVM (Attisano et al. 1993).
The second locus, HHT-2, harbors the activin-like kinase receptor-1 (ALK-1) gene, which has been mapped to the pericentromeric region of chromosome 12 (Vincent et al. 1995; Johnson et al. 1996). The ALK-1 protein has the properties of a type I serine-threonine kinase receptor (Attisano et al. 1993; ten Dijke et al. 1993). Both genes are members of the TGF-β receptor superfamily (Cheifetz et al. 1992; Attisano et al. 1993; ten Dijke et al. 1993) and are highly expressed in endothelial cells and other vascularized tissues such as lung and placenta (Attisano et al. 1993).
The ALK-1 gene is a type I cell surface receptor for the transforming growth factor β (TGF-β) and has the properties of a type I serine threonine kinase receptor superfamily of ligands; however, its ligand and corresponding type II receptor are unknown. This receptor has been shown to bind either activin or TGF-β in the presence of their respective type II receptors, but it does not bind to any ligand alone (Attisano et al. 1993). These genes mediate binding and signaling of TGF-β. ALK-1 can interact with TGF-β or activin, type II receptors as shown by Oh et al. 2000, but the mechanisms for downstream signaling of ALK-1 are yet to be elucidated.
Mutations in the ALK-1 gene in HHT-2 patients include nonsense and missense mutations, as well as insertions and deletions resulting in frameshifts (Johnson et al. 1996; Piantanida et al. 1996; Olivieri et al. 2002).
These mutations have been found throughout the ALK-1 gene coding region, including the extracellular ligand binding, transmembrane and the intracellular kinase domains. The nature and location of these mutations suggest that HHT type 2 results from loss of function of the ALK-1 gene.
In order to analyze the molecular abnormalities in ten unrelated patients with HHT, we have performed a complete sequencing of the coding region of the ENG and activin genes. We were able to identify mutations only in the ALK-1 gene in five patients.
We have developed polymerase chain reaction (PCR) assays to amplify each exon of the coding region of the ALK-1 and ENG gene from genomic DNA. The ALK-1 and ENG genes were sequenced in ten related and unrelated patients with HHT.
Subjects, materials and methods
Human subjects
Informed consent was obtained from all patients and controls, and the study was approved by the local ethics committee.
A diagnosis of HHT was carried out in ten unrelated patients by (1) family history, (2) recurrent episodes of bleeding, and (3) the presence of multiple telangiectatic lesions (Shovlin et al. 2000). The summary of the patients’ clinical data is shown in Table 1. Vascular abnormalities in the gastrointestinal tract, pulmonary, hepatic, and brain circulations were investigated by at least one objective diagnostic method such as endoscopy, ultrasound, and computerized tomography. A panel of 252 chromosomes from unrelated individuals without the disease was used to evaluate the frequency of each mutation in the general population.
Materials and methods
Extraction of DNA from peripheral blood
Genomic DNA was extracted from 1–3 ml of peripheral blood lymphocytes and separated from blood collected in EDTA obtained from healthy volunteers and individuals with HHT in a steady state, using standard procedures.
Mutation analysis
Initially, PCR and sequencing were performed, and when frameshift was detected, this fragment was then cloned and sequenced.
PCR amplification of individual exons
All coding regions and flanking intronic sequences of the ENG and ALK-1 genes were amplified by PCR. Primer sequences and conditions for both genes were previously described by Berg et al. (1997) and McAllister et al. (1994a, b), respectively. PCR products were run on 1.5% agarose/0.5 TBE gels.
Cloning of the PCR products
Polymerase chain reaction products were cloned into the commercial vector SureClone Ligation kit (Amersham Biosciences, San Francisco, CA), and plasmid DNA was prepared using the Wizard Plus SV Minipreps kit (Promega; http://www.promega.com). Cycle sequencing was performed using the MegaBACE Dye Terminator procedure, and reactions were analyzed by a MegaBACE 1000 automatic sequencer (GE Healthcare Life Sciences, USA; http://www.gehealthcare.com). Conservation of the region affected by the mutation was determined by the use of the Blast algorithm to match the ALK-1 region containing the mutation against the GenBank (http://www.ncbi.nlm.nih.gov) nonredundant protein database.
Automized sequencing
Direct sequencing of PCR plasmid products were sequenced in forward and reverse orientation on a MEGABACE 1000 sequencer using the DYEnamic ET Terminator cycle sequencing kit (GE Healthcare, USA; http://www.gehealthcare.com), according to the manufacturer’s instructions. The sequences were analyzed by the sequence analyzed software using the Base Caller Cimarron 3.12 (GE Healthcare, USA; http://www.gehealthcare.com).
Polymorphism screening
For each mutation, a panel of 126 healthy normal control individuals' genomic DNAs from unrelated individuals was screened. As a screening method for mutation detection, we carried out conformation-sensitive gel electrophoresis (CSGE) as described by Ganguly et al. (1993), a method that allows the detection of single base mismatches in DNA heteroduplexes. We have utilized CSGE to screen for potential mutations in a mixed population of normal controls. The entire coding region and intron–exon boundaries of the ALK-1 gene and ENG gene were analyzed.
Following the amplification of the PCR product, 20 μl of each sample was heated at 98°C for 5 min followed by incubation at 68°C for 1 h to generate heteroduplexes. The best separations of heteroduplexes and homoduplexes were obtained with a standard 6% polyacrylamide gel polymerized in 10% ethyleneglycol/15% formamide/Tris-taurine buffer 0.5% (Sigma-Aldrich Brasil Ltd; http://www.sigma-aldrich.com.br).
Results
Identification of novel mutations detected in the ALK-1 gene
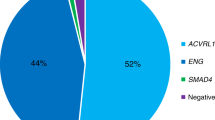
In this article, we studied ten patients with clinical features of HHT; five of them presented four mutations in the ALK-1 gene (three novel and one already described). ENG was not found in any of the ten analyzed patients.
As to the three novel mutations in the ALK-1 gene, two of them are frameshift mutations identified in exons 3 and 7 after direct sequencing of the PCR products, suggesting the existence of a deletion or insertion, and one is a missense mutation identified in exon 7. As to the mutation already described in the literature, it is a missense identified in exon 8.
The first novel mutation was a T deletion (c.913delT) in exon 7 in three patients (brother/sister and one unrelated identified at Table 2 and Fig. 1 as F3 and F2, respectively). The same patient (brother, F3) also presented the second missense mutation (c.976A > G), which was not found in his sister (F3).

Pedigrees of five HHT families: affected individuals are indicated by black symbols and unaffected by white symbols. Squares represented males, circles represent females, and a slashed symbol indicates a deceased individual. The individuals of the families F1 (I-1 and II-1), F2 (II-1), F3 (III-5 and III-6), F4 (II-5 and III-1), and F5 (III-1) had all been sequenced once, and joined mutations are in Table 2. The other individuals of F3 (I-1, II-1, II-6 and III-2), F4 (I-2, II-4 and III-5), and F5 (I-1,II-1, II-2 and II-3) had not been sequenced and had only been cited by the patients as carrying the illness
The third novel mutation (c.204insG) was a G insertion in the exon 3 (father and son: Table 2; Fig. 2 as F1).

The diagrammatic representation of the ALK-1 gene. The exons are marked by boxesunblackened for the coding regions and blackened for the noncoding regions. The ALK-1 sequence alterations are identified during all the length of the gene and include missense and frameshift sequence changes
The fourth mutation, which is a missense and has already been described in the literature (c.1204G > A), was found in three patients (aunt/niece and one unrelated, Table 2 and Fig. 1 as F4 and F5, respectively; Abdalla et al. 2000; Trembath 2001; Olivieri et al. 2002; Lesca et al. 2004).
Those four mutations identified in our five patients are shown in Table 2 and Fig. 2. For each mutation a screening in a panel of 126 normal control individuals was performed to define if these mutations would not represent genetic polymorphisms of the studied population. The summary of the mutations identified is shown in Table 2 and Fig. 2.
Discussion
Endoglin and ALK-1 have been reported to demonstrate an association with HHT (McAllister et al. 1994a, b), an autossomal dominant disorder that causes localized angiodysplasia (Johnson et al. 1996).
Phenotypic penetrance in HTT is age dependent and, at about the age of 40 years, patients are expected to present characteristic clinical manifestations. The inheritance of at least one single mutant copy can predispose the individual to the development of the vascular abnormalities observed in HHT, which are modulated by several genetic, physiologic, and mechanical events (Porteous et al. 1992). Little is known about the genetic basis of the observed clinical heterogeneity of HHT. Even within the same family, there could be great variations with respect to the manifestation and severity of the disease (Shovlin et al. 1997).
These data indicate that loss-of-function mutations in a single allele of the ALK1 locus are sufficient to contribute to defects in maintaining endothelial integrity.
In our work with this series of patients with type 2 HHT, three novel mutations and a mutation already described in the ALK-1 gene were identified. All of these four mutations affect the kinase domain of the ALK-1 gene, and both the deletion and the insertion change conserved amino acids.
From the four detected ALK-1 gene mutations, the insertion within exon 3 encodes the extracellular domain. The T deletion within exon 7 and the other two mutations within exon 7 and 8 encode the kinase domain located near highly conserved cysteines. Several studies suggest that the majority of the mutations grossly truncate the ALK-1 protein and are, thus, classic functional null alleles (McAllister et al. 1994a, b; Johnson et al. 1996; Gallione et al. 1998; Klaus et al. 1998; Pece-Barbara et al. 2005).
Five patients did not present any mutations in ALK1 or ENG genes, suggesting that the disease may be linked to other loci. In these five patients, the ENG gene was sequenced, and no mutation was found. We have performed the whole coding sequences of both genes (ALK-1 and ENG), suggesting the existence of a third HHT gene mentioned by Piantanida et al. (1996). Wallace and Shovlin (2000) confirmed, by linkage analyses, the exclusion of linkage to ENG and activin (chromosome 9 and chromosome 12).
Indeed, the presence of a third rare variant may be speculated, as suggested by Piantanida et al. (1996) and Wallace and Shovlin (2000), who described the existence of another locus accounting for the disease in some HHT patients with pulmonary involvement.
The existence of another locus accounting for disease in some HHT patients with hepatic involvement was described by Cole et al. (2005), a new locus for HHT3 has been mapped at chromosome 5 (5q31.3–5q32) and will probably account for a subset of individuals without mutations in the ENG or ALK-1 genes.
One of the central questions in HHT research has been the cause of the disease phenotype, whether it is due to haploinsufficiency or to dominant-negative interactions between mutant and normal proteins. The current model, therefore, is that the disease phenotype is the result of inherited haploinsufficiency of either ALK-1 or ENG genes. Missense mutations that are stably expressed can result in constitutively active proteins or in a gain of function, or they can display a dominant-negative mechanism (Wilkie 1994).
We describe herein three novel mutations in the ALK-1 gene in a group of Brazilian HHT patients. All of the mutations detected are novel and have not been published previously. From an initial group of ten patients, we identified ALK-1 mutations in five of them, as demonstrated in Table 2 and Fig. 2. As it was not possible to establish specific ALK-1 gene mutations in the other five patients, we performed a genetic study in the ten patients, and no ENG mutations were identified, suggesting that the phenotypic pattern of HTT may be linked to distinct genetic regulators, such as mutations in other genes or alterations in the promoter region. Since, to date, no mutations in the ALK-1 exon 1 have been reported, this region was not included in the analysis as described by Attisano et al. (1993), the exon I contains the 5′ untranslated sequence and is part of exon II; for ten Dijke et al. (1993), it is a splice variant that appears from the splicing of exon I to a consensus splice junction present 7 bp upstream of the start in exon II. In five alleles, we found frameshift mutations such as a G insertion within the exon 3 (Table 2, Fig. 1 as F1; which encodes the extracellular domain) and the deletion of a single T nucleotide in exon 7 (Table 2, Fig. 1 as F3; a kinase domain). Two missense mutations were also characterized in a highly conserved cysteine-containing region, related to exons 7 and 8: a p.Ile326Val substitution and a p.Gly402Ser substitution (Table 2, Fig. 1 as F4 and F5), respectively.
In one patient, it was possible to characterize the co-inheritance of both the T deletion (p.Ser305fs) and the p.Ile326Val substitution (Fig. 1 as F3). To the best of our knowledge, this is the first description of two mutant alleles in the same individual. One important question is concerned about the genotypic determinants of HHT phenotypes. Commonly, TGF-β may activate both the ALK-1 and ENG in association with the receptor II. One possible explanation for the HTT phenotype variability may be the existence of an abnormal ALK1 protein, which might have the ability to bind to the type II receptor without producing signal transduction, either because of kinase domain disruption or due to extracellular domain alteration, leading to ligand-binding failure (Piantanida et al. 1996).
The lack of correlation between the genotype and clinical manifestations of HHT supports the haploinsufficiency model and dominant-negative effect as suggested in McAllister et al. (1995) and Berg et al. (1997). This current model postulates that the disease phenotype may be developed despite the presence of at least a half normal tissue ALK-1 or ENG, which indicates a dominant-negative mechanism (Wilkie 1994; Pece-Barbara et al. 2005).
Several studies have suggested that pulmonary AVMs are more frequent in HHT1 than HHT2 patients.
Our results support the hypothesis that HHT2, like HHT1, is associated with haploinsufficiency, as previously suggested (Piantanida et al. 1996).
We conclude that the rate of mutation detection by the genomic sequence of the ALK-1 gene suggests that this will be a potential useful diagnostic test for HHT type 2. Moreover, this approach will be particularly important for identifying young HHT patients at high risk of developing severe complications.
References
Abdalla SA, Pece-Barbara N, Vera S, Tapia E, Paez E, Bernabeu C, Letarte M (2000) Analysis of ALK-1 and endoglin in newborns from families with hereditary haemorrhagic telangiectasia type 2. Hum Mol Genet 9(8):1227–1237
Attisano L, Carcamo J, Ventura F, Weis FM, Massague J, Wrana JL (1993) Identification of human activin and TGF beta type I receptors that form heteromeric kinase complexes with type II receptors. Cell 75(4):671–680
Berg JN, Guttmacher AE, Marchuk DA, Porteous ME (1996) Clinical heterogeneity in hereditary haemorrhagic telangiectasia: are pulmonary arteriovenous malformations more common in families linked to endoglin? J Med Genet 33(3):256–257
Berg JN, Gallione CJ, Stenzel TT, Johnson DW, Allen WP, Schwartz CE, Jackson CE, Porteous ME, Marchuk DA (1997) The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary haemorrhagic telangiectasia type 2. Am J Hum Genet 61(1):60–67
Berg J, Porteous M, Reinhardt D, Gallione C, Holloway S, Umasunthar T, Lux A, McKinnon W, Marchuk D, Guttmacher A (2003) Hereditary haemorrhagic telangiectasia: a questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. J Med Genet 40(8):585–590
Cheifetz S, Bellon T, Cales C, Vera S, Bernabeu C, Massague J, Letarte M (1992) Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J Biol Chem 267(27):19027–19030
Cole SG, Begbie ME, Wallace GM, Shovlin CL (2005) A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet 42(7):577–582
Gallione CJ, Klaus DJ, Yeh EY, Stenzel TT, Xue Y, Anthony KB, McAllister KA, Baldwin MA, Berg JN, Lux A, Smith JD, Vary CP, Craigen WJ, Westermann CJ, Warner ML, Miller YE, Jackson CE, Guttmacher AE, Marchuk DA (1998) Mutation and expression analysis of the endoglin gene in hereditary haemorrhagic telangiectasia reveals null alleles. Hum Mutat 11(4):286–294
Ganguly A, Rock MJ, Prockop DJ (1993) Conformation-sensitive gel electrophoresis for rapid detection of single-base differences in double-stranded PCR products and DNA fragments: evidence for solvent-induced bends in DNA heteroduplexes. Proc Natl Acad Sci USA 90(21):10325–10329
Gougos A, Letarte M (1990) Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J Biol Chem 265(15):8361–8364
Guttmacher AE, Marchuk DA, White RI Jr (1995) Hereditary haemorrhagic telangiectasia. N Engl J Med 333(14):918–924
Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME, Marchuk DA (1996) Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 13(2):189–195
Kjeldsen AD, Vase P, Green A (1999) Hereditary haemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. J Intern Med 245(1):31–39
Klaus DJ, Gallione CJ, Anthony K, Yeh EY, Yu J, Lux A, Johnson DW, Marchuk DA (1998) Novel missense and frameshift mutations in the activin receptor-like kinase-1 gene in hereditary haemorrhagic telangiectasia. Mutations in brief no. 164. Online. Hum Mutat 12(2):137
Lesca G, Plauchu H, Coulet F, Lefebvre S, Plessis G, Odent S, Riviere S, Leheup B, Goizet C, Carette MF, Cordier JF, Pinson S, Soubrier F, Calender A, Giraud S (2004) Molecular screening of ALK1/ACVRL1 and ENG genes in hereditary haemorrhagic telangiectasia in France. Hum Mutat 23(4):289–299
McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J et al (1994a) Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 8(4):345–351
McAllister KA, Lennon F, Bowles-Biesecker B, McKinnon WC, Helmbold EA, Markel DS, Jackson CE, Guttmacher AE, Pericak-Vance MA, Marchuk DA (1994b) Genetic heterogeneity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 31(12):927–932
McAllister KA, Baldwin MA, Thukkani AK, Gallione CJ, Berg JN, Porteous ME, Guttmacher AE, Marchuk DA (1995) Six novel mutations in the endoglin gene in hereditary haemorrhagic telangiectasia type 1 suggest a dominant-negative effect of receptor function. Hum Mol Genet 4:1983–1985
Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, Li E (2000) Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA 97(6):2626–2631
Olivieri C, Mira E, Delu G, Pagella F, Zambelli A, Malvezzi L, Buscarini E, Danesino C (2002) Identification of 13 new mutations in the ACVRL1 gene in a group of 52 unselected Italian patients affected by hereditary haemorrhagic telangiectasia. J Med Genet 39(7):E39
Pece-Barbara N, Vera S, Kathirkamathamby K, Liebner S, Di Guglielmo GM, Dejana E, Wrana JL, Letarte M (2005) Endoglin null endothelial cells proliferate faster and are more responsive to transforming growth factor beta1 with higher affinity receptors and an activated Alk1 pathway. J Biol Chem 280(30):27800–27808
Piantanida M, Buscarini E, Dellavecchia C, Minelli A, Rossi A, Buscarini L, Danesino C (1996) Hereditary haemorrhagic telangiectasia with extensive liver involvement is not caused by either HHT1 or HHT2. J Med Genet 33(6):441–443
Plauchu H, de Chadarevian JP, Bideau A, Robert JM (1989) Age-related clinical profile of hereditary haemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 32(3):291–297
Porteous ME, Burn J, Proctor SJ (1992) Hereditary haemorrhagic telangiectasia: a clinical analysis. J Med Genet 29(8):527–530
Sawabe M, Arai T, Esaki Y, Tsuru M, Fukazawa T, Takubo K (2001) Three-dimensional organization of the hepatic microvasculature in hereditary haemorrhagic telangiectasia. Arch Pathol Lab Med 125(9):1219–1223
Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, Kjeldsen AD, Plauchu H (2000) Diagnostic criteria for hereditary haemorrhagic telangiectasia (Rendu–Osler–Weber syndrome). Am J Med Genet 91(1):66–67
Shovlin CL, Hughes JM, Scott J, Seidman CE, Seidman JG (1997) Characterization of endoglin and identification of novel mutations in hereditary haemorrhagic telangiectasia. Am J Hum Genet 61(1):68–79
ten Dijke P, Ichijo H, Franzen P, Schulz P, Saras J, Toyoshima H, Heldin CH, Miyazono K (1993) Activin receptor-like kinases: a novel subclass of cell-surface receptors with predicted serine/threonine kinase activity. Oncogene 8(10):2879–2887
Trembath RC (2001) Mutations in the TGF-beta type 1 receptor, ALK1, in combined primary pulmonary hypertension and hereditary haemorrhagic telangiectasia, implies pathway specificity. J Heart Lung Transplant 20(2):175
Vincent P, Plauchu H, Hazan J, Faure S, Weissenbach J, Godet J (1995) A third locus for hereditary haemorrhagic telangiectasia maps to chromosome 12q. Hum Mol Genet 4(5):945–949
Wallace GM, Shovlin CL (2000) A hereditary haemorrhagic telangiectasia family with pulmonary involvement is unlinked to the known HHT genes, endoglin and ALK-1. Thorax 55(8):685–690
Wilkie AO (1994) The molecular basis of genetic dominance. J Med Genet 31(2):89–98
Acknowledgments
We thank Vera Suzigan for proofreading the manuscript. This work was supported by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) and FAPESP (Fundação de Amparo à pesquisa do Estado de São Paulo).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Assis, A.M., Costa, F.F., Arruda, V.R. et al. Three novel mutations in the activin receptor-like kinase 1 (ALK-1) gene in hereditary hemorrhagic telangiectasia type 2 in Brazilian patients. J Hum Genet 52, 237–243 (2007). https://doi.org/10.1007/s10038-006-0104-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0104-3
