
Abstract
Patterns of linkage disequilibrium (LD) act as the framework for designing efficient association studies; these patterns are being studied and catalogued by The International HapMap Project. The current study assessed the transferability of tag SNPs chosen from HapMap panels to a cohort of 80 individuals from metro Cebu, Philippines, who participated in the Cebu Longitudinal Health and Nutrition Survey (CLHNS). The analyses focused on 627 single nucleotide polymorphisms (SNPs) in the central 40 kb within each of the 10 HapMap ENCODE regions. The similarity between the genetic variants in Cebu Filipino samples and HapMap panels was examined using allele frequency estimates, measures of pairwise linkage disequilibrium (LD), and haplotype frequency estimates. For these measures, strong correlations were observed between the Cebu Filipino samples and the Asian panels from HapMap, with the strongest correlations observed with the Han Chinese from Beijing (CHB) panel. Tag SNPs selected using the HapMap CHB panel were particularly effective at representing the genetic variation in Cebu Filipino samples. These results suggest that the HapMap data will be an effective resource for future studies in Cebu Filipino samples.
Similar content being viewed by others

Introduction
Genetic association studies make use of patterns of linkage disequilibrium (LD) between genetic polymorphisms for efficient genotyping (Carlson et al. 2004; Service et al. 2006). LD patterns reflect the ancestry of a population and vary considerably throughout the genome (Reich et al. 2001; Sawyer et al. 2005; Plagnol and Wall 2006). The International HapMap Consortium is studying and cataloguing DNA sequence variation and characterizing these patterns of LD across the genome (The International HapMap Consortium 2005), allowing correlated single nucleotide polymorphisms (SNPs) to be excluded from genotyping, but considered in analysis. The HapMap is made up of four panels of dense haplotype maps for individuals characterized as Han Chinese from Beijing, China (CHB), Japanese from Tokyo, Japan (JPT), Caucasians from Utah, USA, with northern and western European ancestry (CEU), and Yoruban of Ibadan, Nigeria (YRI).
The HapMap is an important resource for choosing tag SNPs in disease association and population studies (The International HapMap Consortium 2005). However, worldwide population variation is not completely characterized, and an essential question is whether tag SNPs chosen using HapMap panels will adequately capture patterns of genetic variation in other populations (Weale et al. 2003; Nejentsev et al. 2004; Evans and Cardon 2005; Ke et al. 2005; Mueller et al. 2005; Ramirez-Soriano et al. 2005; Gonzalez-Neira et al. 2006; Huang et al. 2006; Montpetit et al. 2006; Ribas et al. 2006; Willer et al. 2006). Furthermore, for populations similar to those genotyped in the HapMap project, HapMap data may be used to directly predict genotypes of non-tag SNPs for analysis in association studies (Eyheramendy et al. 2007; Paschou et al. 2007). Previous studies observed that the HapMap CHB and JPT panels have very similar patterns of LD and could act as a proxy for other geographically related populations (Beaty et al. 2005; Lim et al. 2006; de Bakker et al. 2006; Conrad et al. 2006; Mahasirimongkol et al. 2006; Yoo et al. 2006). However, data are currently unavailable to assess the effectiveness of using the existing HapMap data to guide SNP selection and interpretation for samples from the Cebu Longitudinal Health and Nutrition Survey (CLHNS) cohort from metro Cebu in the central Philippines (Cebu Study Team 1991; Adair 2004). This study assesses the advantages of having HapMap data from two related Asian panels to evaluate whether the combined CHB and JPT panels would more effectively capture genetic variability in Cebu Filipinos than the CHB or JPT panels alone. In addition, this study develops the most efficient criteria for selecting tag SNPs from the HapMap panels for future genetic association studies in Cebu Filipino samples.
To address these issues, SNPs from within the ten HapMap ENCyclopedia Of DNA Elements (ENCODE) reference regions were used (The ENCODE Project Consortium 2004). These regions were re-sequenced in 48 unrelated individuals (8 CHB, 8 JPT, 16 CEU, and 16 YRI) for SNP discovery and reflect the density of SNPs in the genome more accurately than other regions in HapMap. The SNP density in these ENCODE regions is higher than the remainder of HapMap (The International HapMap Consortium 2005). The similarity of the HapMap samples to 80 Cebu Filipino samples was assessed, using allele frequency estimates, pairwise LD (r 2), and haplotype frequency estimates as measures of similarity. Furthermore, the efficiency of using tag SNPs selected from the HapMap Asian panels for capturing genetic variation in Cebu Filipino samples was studied.
Materials and methods
Samples
Eighty unrelated Cebu Filipino individuals were randomly selected from a cohort of healthy women from the CLHNS (www.cpc.unc.edu/projects/cebu). Informed consent was obtained from all individuals and the study protocol was approved by the University of North Carolina Institutional Review Board for the Protection of Human Subjects.
Genomic DNA was isolated from peripheral blood lymphocytes using automated and manual DNA extraction methods (Puregene, Gentra) by the University of North Carolina, Chapel Hill BioSpecimen Processing Facility. Centre d’Etude du Polymorphisme Humain (CEPH) DNA samples were obtained from Coriell (Camden, NJ).
HapMap genotype data were obtained from the HapMap database (www.hapmap.org) for all available unrelated individuals, including 45 CHB, 44 JPT, 60 CEU parents of trios, and 60 YRI parents of trios. For some analysis, the CHB and JPT samples were combined (indicated as CHB + JPT) (The International HapMap Consortium 2005).
SNP selection and genotyping
To represent the overall complexity of the genome, the central 40-kb region from within each of the ten 500-kb ENCODE regions that have been used for SNP discovery and dense SNP genotyping was chosen for this study (Table 1). SNPs were selected if they were polymorphic (minor allele frequency, MAF, >0) in the HapMap CHB, JPT, or CEU panels.
Of the 883 SNPs that met these criteria, 215 were eliminated based on Illumina design score (calculated December 2005). One SNP identified by re-sequencing region ENr213 in Cebu Filipino samples (see below) was included, resulting in a total of 669 SNPs that were genotyped in the Cebu Filipino samples. SNP genotyping was performed at the Mammalian Genotyping Core at the University of North Carolina, Chapel Hill, using the Illumina GoldenGate (Illumina Inc., San Diego, CA) genotyping assay (Gunderson et al. 2004). Of the 669 SNPs attempted, 36 SNPs were excluded based on poorly defined clusters (n = 28), genotyping completeness <90% (n = 3), or inconsistency with Hardy-Weinberg equilibrium (p < 0.001; n = 5). Six additional SNPs were excluded because of two or more genotype discrepancies between six CEPH DNA samples and equivalent HapMap CEU genotypes. SNPs were also evaluated for two or more genotyping discrepancies between seven duplicate samples; however, no SNPs needed to be dropped based on this criterion. The genotyping success rate of the final 627 SNPs was 99.9%, and the discrepancy rate was 0.02%. Of these 627 SNPs, 501 (80%) were polymorphic (MAF >0) in Cebu Filipino samples. The average marker spacing of these 501 polymorphic SNPs was 1 SNP/798 bp.
Statistical analysis
Tests for consistency of genotype distributions with expected Hardy-Weinberg equilibrium proportions were calculated using standard Pearson’s χ 2 statistics. Only markers with a MAF ≥0.05 in HapMap panels were analyzed in Cebu Filipino samples. SNPs were matched for the reference allele between all HapMap panels and Cebu Filipino samples. Fisher’s exact tests were used to test for allele frequency differences between pairs of samples. Pair-wise LD (r 2) values were calculated using Haploview (Barrett et al. 2005; http://www.broad.mit.edu/mpg/haploview) for adjacent pairs and all pairs of SNPs in each region. Haplotype blocks were defined in Haploview for each HapMap panel based on the default block definition (Gabriel et al. 2002). Identical blocks from each HapMap panel were defined in the Cebu Filipino samples for comparison. Using Haploview, haplotype frequencies were estimated in each haplotype block for every population. Haplotypes with a frequency >0.01 were evaluated in the HapMap panels. Haplotypes not observed in the Cebu Filipino samples were assigned a frequency of zero. Spearman’s correlation coefficients were calculated for all comparisons between Cebu Filipino samples and HapMap panels.
In order to evaluate the efficiency of HapMap to choose tag SNPs for Cebu Filipino samples, tag SNPs from HapMap panels were selected using Tagger in pairwise tagging mode with other settings at default values (de Bakker et al. 2005; http://www.broad.mit.edu/mpg/tagger/). Several r 2 thresholds were used to assess the performance of selecting tag SNPs using the HapMap panels: 0.80, 0.85, 0.90, and 0.95. If a Cebu Filipino SNP exhibited pairwise r 2 ≥0.80 with at least one tag SNP (selected from the Asian HapMap panels), then the SNP was defined as captured in the Cebu Filipino sample. Percent coverage for a region is defined as the number of captured SNPs in the Cebu Filipino samples divided by the total number of SNPs (with estimated MAF ≥0.05). Finally, for each Cebu Filipino SNP, the maximum r 2 estimate obtained over all r 2 estimates between that SNP and a tag SNP in the region was identified. For a region, mean maximum r 2 was defined as the average value of the maximum r 2 values obtained over all Cebu Filipino SNPs in the regions.
Re-sequencing
Twenty-four randomly chosen Cebu Filipino samples were re-sequenced in the central 800 nucleotide (nt) region within each of the 40-kb ENCODE regions. Primers were selected using Primer3 software (Rozen and Skaletsky 2000; http://primer3.sourcegorge.net), and sequences were compared using Sequencher 4.2.2 (Gene Codes Corporation, Ann Arbor, MI). Sequencing was performed at the University of North Carolina, Chapel Hill, automated DNA sequencing facility on an ABI Prism 3730 (Applied Biosystems, Foster City, CA) using the Big Dye Terminator Kit.
Results
To determine the extent of similarity between 80 Cebu Filipino samples and HapMap samples, genotype data for 627 SNPs located within the ten HapMap ENCODE regions was used (Table 1).
Allele frequencies
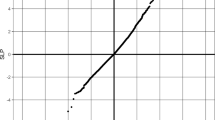
Allele frequency estimates were compared using SNPs with MAF ≥0.05 in the corresponding HapMap panel. A total of 399 SNPs were evaluated when examining CHB, 391 SNPs for JPT, 396 SNPs for CHB+JPT, 431 SNPs for CEU, and 391 SNPs for YRI. The Spearman’s correlation coefficients for allele frequency estimates between the Cebu Filipino samples and the HapMap panels were 0.96, 0.92, 0.95, 0.82, and 0.65 for CHB, JPT, CHB+JPT, CEU, and YRI, respectively (Fig. 1). For comparison, the Spearman’s correlation coefficient for allele frequency estimates between CHB and JPT samples was 0.95 for 384 SNPs with MAF ≥0.05 in both panels. The percent of SNPs with significantly different allele frequencies (Fisher’s exact p-value < 0.01) was 5.7% for CHB, 15.6% for JPT, 11.6% for CHB + JPT, 57.7% for CEU, and 60.1% for YRI. Although larger sample sizes should provide greater power to detect statistically significant differences, the 89 CHB + JPT samples showed fewer significant differences with the Cebu Filipino samples than the smaller JPT, CEU, or YRI groups. The allele frequency comparison was repeated using HapMap SNPs with MAF >0 and slightly higher Spearman’s correlations were obtained with analogous patterns of similarity (data not shown).

Comparison of allele frequency estimates between Cebu Filipino samples and HapMap samples for SNPs with MAF ≥0.05 in the HapMap sample. Open symbols indicate SNPs with significantly different allele frequencies at a Fisher’s exact p-value <0.01
Based on the substantially greater similarity in allele frequencies between Cebu Filipino samples and Asian HapMap panels compared to CEU or YRI panels, subsequent analyses were performed using only the HapMap CHB, JPT, and CHB + JPT panels.
Linkage disequilibrium
Pairwise r2 for adjacent pairs and all pairs of SNPs within each HapMap ENCODE region in the Asian HapMap samples and Cebu Filipino samples were estimated to evaluate the extent of LD in each population. Only SNPs with MAF ≥0.05 in the corresponding HapMap sample were included in comparisons. Analysis was performed for 375, 368, and 373 adjacent pairs of SNPs and 9,350, 8,912, and 9,157 total pairs of SNPs for CHB, JPT, and CHB + JPT, respectively. The Spearman’s correlation coefficients of the r 2 estimates for adjacent pairs of SNPs between the Cebu Filipino and Asian HapMap samples were 0.90 for each of CHB, JPT, and CHB + JPT. The Spearman’s correlation coefficients of the r 2 estimates for all pairs were 0.88 for CHB, 0.87 for JPT, and 0.89 for CHB + JPT (Table 2). The absolute difference between r 2 estimates of adjacent SNP pairs was calculated. For CHB 51, 73, and 85% of the SNPs had absolute differences between r 2 estimates of ≤0.05, ≤0.10, and ≤0.15; for JPT 48, 67, and 80% of the SNPs had absolute differences between r 2 estimates of ≤0.05, ≤0.10, and ≤0.15; for CHB + JPT 50, 69, and 83% of the SNPs had absolute differences between r 2 estimates of ≤0.05, ≤0.10, and ≤0.15.
When each of the ten regions was analyzed separately, LD differed both among regions and populations. Region ENr232 varied the most between HapMap panels; Spearman’s correlation coefficients of the r 2 estimates, for all pairs of SNPs, between the Cebu Filipino and Asian HapMap samples were 0.85 for CHB, 0.63 for JPT, and 0.77 for CHB + JPT. This region, however, did not differ from the other regions in allele frequency estimates, haplotype frequency estimates (below), and tag SNP analyses (below). The pairwise r 2 analysis was repeated using all HapMap SNPs with MAF >0 and obtained slightly higher Spearman’s correlations, but analogous patterns of similarity (data not shown). To confirm that Cebu Filipino sample size did not impact results, the analysis was repeated with three random sets of 45 Cebu Filipino samples. The sets of 45 were compared to CHB and JPT panels, and similar results were observed for the total set of 80 Cebu Filipino samples (data not shown). On average across all regions, Cebu Filipino samples show highly similar patterns of LD compared to all Asian panels, with slightly more similarity observed with CHB + JPT panels and slightly less observed with JPT panels.
Haplotype frequencies
Haplotype frequencies for the Asian HapMap panels and Cebu Filipino samples were estimated for haplotypes comprised of SNPs with MAF ≥0.05 in the HapMap panel. Haplotype blocks were defined using the default block definition used in Haploview (Gabriel et al. 2002). Within the ten regions, the average number of blocks per region was 3.6, 3.6, and 3.3 for CHB, JPT and CHB + JPT, respectively. The blocks ranged in size from 2 to 65 SNPs with an average of 9.7, 10.9, and 9.1 SNPs per block for CHB, JPT, and CHB + JPT. One hundred seventy-eight, 151, and 141 haplotypes were identified with frequency estimates >0.01 in CHB, JPT, and CHB + JPT, respectively. The Spearman’s correlation coefficient of haplotype frequency estimates between Cebu Filipino and Asian HapMap samples was 0.95 for CHB, 0.88 for JPT, and 0.92 for CHB+JPT (Fig. 2). Most haplotypes with an estimated frequency >0 in the Asian samples were also observed (with estimated frequency >0) in Cebu Filipino samples, demonstrating a high degree of haplotype conservation across the populations. Of the observed haplotypes with estimated frequency ≥0.05 in CHB, JPT, and CHB + JPT, only 2.5% (3 of 119), 2.8% (3 of 107), and 1.8% (2 of 112), respectively, were not observed in Cebu Filipino samples. In addition, of the observed haplotypes with estimated frequency >0.01 in CHB, JPT, and CHB+JPT, 23% (41 of 178), 24% (36 of 151), and 11% (16 of 141), respectively, were not observed in Cebu Filipino samples. The greater representation of Cebu Filipino haplotypes in CHB + JPT samples is likely attributed to the larger sample size. Overall, the haplotype frequency differences were modest between Cebu Filipino samples and the Asian HapMap panels, with CHB showing the most similarity and JPT showing the least similarity.

Comparison of haplotype frequency estimates between Cebu Filipino samples and Asian HapMap samples for SNPs with MAF ≥0.05 in the HapMap sample and haplotype frequency estimates >0.01 in the HapMap sample
Transferability of tag SNPs
To measure the efficiency of using the HapMap panels for tag SNP selection in the Cebu Filipino population, Tagger was used to select tag SNPs from the CHB, JPT, and CHB + JPT panels for SNPs with MAF ≥0.05. Tag SNP coverage was tested at four r 2 thresholds for selection in the HapMap panels, and the tag SNPs chosen in HapMap panels were applied to SNPs with MAF ≥0.05 in Cebu Filipino samples.
Overall, at each r 2 selection threshold using the CHB, JPT, and CHB + JPT panels, the percentages of SNPs captured (with a mean r 2 ≥ 0.80) in the Cebu Filipino samples were very similar. Using any of the three panels for SNP selection, the lowest r 2 selection threshold of 0.80 captured at least 82–83% of Cebu Filipino SNPs (MAF ≥0.05) across all ten regions (Table 3). To obtain this percent coverage 121, 118, and 125 tag SNPs from CHB, JPT, and CHB + JPT, respectively, would need to be genotyped. As expected, increasing the r 2 threshold for selecting tag SNPs in the Asian HapMap samples increased both the number of tag SNPs that needed to be genotyped and the proportion of Cebu Filipino SNPs captured by these tag SNPs. However, the percent coverage of each region varied substantially. At the r 2 selection threshold of 0.80, the percent coverage ranged over the ten regions from 54 to 96%, 59 to 96%, and 52 to 94% using CHB, JPT, and CHB + JPT tag SNPs, respectively. This variability between regions was still observed at a r 2 selection threshold of 0.95, the highest r 2 threshold studied. In addition, at each r 2 selection threshold, the mean maximum r 2 of all Cebu Filipino SNPs (MAF ≥0.05) was similar between CHB, JPT, and CHB + JPT. Among all SNPs, for the r 2 selection threshold of 0.80, a mean maximum r 2 of 0.88 was observed using CHB, JPT, and CHB + JPT tag SNPs. As expected, the mean maximum r 2 increased at each increase of the r 2 selection threshold. Little variability was observed between regions (data not shown).
At each r 2 selection threshold, the SNPs in the Cebu Filipino samples that were not captured by a tag SNP selected in the HapMap panels were evaluated. The percentage of SNPs not captured and the mean maximum r 2 for each SNP were calculated (Table 3). Consistent with the sensitivity of tag SNP selection to allele frequency (Schulze et al. 2004), many of the SNPs not captured were rare (MAF <0.10). These rare SNPs had low mean maximum r 2 and were not captured using higher r 2 selection thresholds. Common SNPs (MAF ≥0.10) that were not captured at an r 2 of at least 0.80 were captured with at least a mean maximum r 2 of 0.65, 0.64, and 0.66 using CHB, JPT, and CHB + JPT tag SNPs, respectively. As the r 2 selection threshold increased, more of these Cebu Filipino common SNPs were captured with an r 2 of at least 0.80.
Re-sequencing
To assess the frequency of population-specific novel SNPs and to further evaluate the genetic structure in Cebu Filipinos, 24 Cebu Filipino individuals were re-sequenced in an 800-nt region within each of the ten HapMap ENCODE regions used previously for HapMap re-sequencing (The ENCODE Project Consortium 2004). Approximately 184 kb on at least one DNA strand were re-sequenced. Only one novel SNP was detected that was not present in HapMap (data release 21, July 2006) or dbSNP (build 126); the SNP was located in region ENr213 (ss69374772) and had a MAF of 0.05 in 80 Cebu Filipino individuals. Within Cebu Filipino samples, this SNP exhibited a maximum r 2 of 0.228 with four other SNPs in the 40-kb region.
Discussion
The extent of similarity between Cebu Filipino samples and the previously evaluated HapMap samples were examined using measures of allele frequency estimates, pairwise r 2 estimates, and haplotype frequency estimates. Consistent with population migration, mitochondrial DNA, and Y haplotype patterns (Jin and Su 2000), CEU and YRI samples were much less similar to Cebu Filipino samples with respect to allele frequency than CHB, JPT, or CHB + JPT samples. All of the analyses showed high similarity between Asian HapMap samples and Cebu Filipino samples.
Because the CHB and JPT samples have similar allele frequencies, these data sets are often combined for analyses (The International HapMap Consortium 2005). The existence of these two Asian HapMap panels allowed for evaluating the choice of using CHB, JPT, or the larger combined CHB + JPT panel as a resource for choosing haplotype tagging SNPs for Cebu Filipino samples. Among these three panels, JPT samples were the least correlated with Cebu Filipino samples with respect to allele frequency estimates, pairwise r 2 estimates, and haplotype frequency estimates. Cebu Filipino and CHB allele frequency estimates were more closely correlated than CHB and JPT allele frequency estimates. Both CHB and CHB + JPT panels were very similar to Cebu Filipino samples, and it is not clear which panel would act most efficiently as a proxy for the Cebu Filipino samples. The larger CHB + JPT sample size would be expected to decrease the variability in the allele and haplotype frequency estimates; the added JPT samples could decrease accuracy. Indeed, estimated Cebu Filipino allele and haplotype frequencies were slightly more correlated with CHB than CHB + JPT, but Cebu Filipino pairwise r 2 estimates were slightly more similar to CHB + JPT than CHB.
A practical use of HapMap is to select tag SNPs for regional or genome-wide association studies (The International HapMap Consortium 2005). Evaluation was performed on the transferability of HapMap tag SNPs chosen using the data from CHB, JPT, and CHB + JPT panels at several r 2 selection thresholds, with respect to capturing the genetic variability in samples from Cebu, Philippines. Using these criteria, at an r 2 selection threshold of 0.80, the HapMap-based tag SNPs capture 82–83% of the Cebu Filipino SNPs. A majority of the most common SNPs (MAF ≥0.10) in the Cebu Filipino sample that are not captured by the tag SNPs at an r 2 of at least 0.80 are captured with an r 2 of at least 0.60. Using higher r 2 thresholds for tag SNP selection in the HapMap samples results in capturing more SNPs in the Cebu Filipino sample, but with the added cost of genotyping more tag SNPs. Increasing the r 2 threshold failed to capture substantially more rare SNPs, most of which exhibited low pairwise LD with other SNPs.
Previously, de Bakker et al. (2006) showed through extensive SNP discovery and simulations that power to detect disequilibrium-based association is only modestly compromised when an appropriate selection of tag SNPs are chosen from HapMap samples and applied to other case-control samples. Large scale SNP discovery and power simulations were beyond the scope of this study. However, based on the findings from de Bakker et al. (2006) and the current findings that tag SNPs selected using the Asian HapMap adequately captured common Cebu Filipino SNPs, the average loss in power to detect common casual alleles should be small.
Re-sequencing and genotyping were performed in the ten HapMap ENCODE regions that were re-sequenced for SNP discovery and are considered to be a gold standard because of the high density of SNP coverage (The ENCODE Project Consortium 2004). Only one SNP (estimated MAF = 0.05) was detected in the Cebu Filipino samples that was not observed in dbSNP or HapMap, suggesting that alleles ascertained from the HapMap ENCODE regions were representative of the common variation in Cebu Filipinos and that additional re-sequencing of these regions would not be required to detect common SNPs in Cebu Filipino samples. While future SNP selection in genome regions that have not been re-sequenced will be based on less complete SNP identification, the Asian HapMap panels will likely either include or tag most of the common SNPs present in Cebu Filipino samples.
Measures of LD, gene density, and haplotype blocks vary across the genome (Ke et al. 2004; De la Vega et al. 2005), and the HapMap ENCODE regions analyzed represent a range of these and other characteristics (The ENCODE Project Consortium 2004), suggesting that our results may apply, on average, across the genome. The strong correlations observed between Cebu Filipino samples and HapMap Asian panels are broadly consistent with other assessments of tagging transferability outside the HapMap ENCODE regions (Weale et al. 2003; Nejentsev et al. 2004; Ke et al. 2005; Mueller et al. 2005; Ramirez-Soriano et al. 2005; Evans and Cardon 2005; Gonzalez-Neira et al. 2006; Huang et al. 2006; Mahasirimongkol et al. 2006; Montpetit et al. 2006; Ribas et al. 2006; Willer et al. 2006).
Our results are consistent with previous studies that compared the Asian HapMap panels to other Eastern Asian samples. Studies that examined many populations worldwide found Asian and Oceania populations to be most similar to the Asian HapMap panel tested (Conrad et al. 2006 and Gonzalez-Neira et al. 2006). Two studies have investigated the tagging transferability between the HapMap CHB, JPT, and CHB + JPT with sample sets from Thailand and from Korea (Mahasirimongkol et al. 2006; Yoo et al. 2006). A combination of tag SNPs from CHB + JPT best captured the LD structure of the Thais, while SNP selection based on JPT was most transferable to the Korean samples. In comparison, our results suggest that CHB samples and the combined CHB + JPT samples are most similar to Cebu Filipino samples, although our results do not necessarily reflect the patterns of genetic variability across the Philippines. Our findings will be useful for the future design and analysis of genetic studies in the Cebu Filipino population.
References
Adair LS (2004) Dramatic rise in overweight and obesity in adult Filipino women and risk of hypertension. Obes Res 12:1335–1341
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Beaty TH, Fallin MD, Hetmanski JB, McIntosh I, Chong SS, Ingersoll R, Sheng X, Chakraborty R, Scott AF (2005) Haplotype diversity in 11 candidate genes across four populations. Genetics 171:259–267
Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA (2004) Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet 74:106–120
Cebu Study Team, Adair L, Akin JS, Black R, Briscoe J, Guilkey DK, Popkin BM, Flieger WF (1991) Underlying and proximate determinants of Child Health: The Cebu Longitudinal Health and Nutrition Study. Am J Epidemiol 133:185–201
Conrad DF, Jakobsson M, Coop G, Wen X, Wall JD, Rosenberg NA, Pritchard JK (2006) A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat Genet 38:1251–1260
de Bakker PI, Burtt NP, Graham RR, Guiducci C, Yelensky R, Drake JA, Bersaglieri T, Penney KL, Butler J, Young S, Onofrio RC, Lyon HN, Stram DO, Haiman CA, Freedman ML, Zhu X, Cooper R, Groop L, Kolonel LN, Henderson BE, Daly MJ, Hirschhorn JN, Altshuler D (2006) Transferability of tag SNPs in genetic association studies in multiple populations. Nat Genet 38:1298–1303
de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D (2005) Efficiency and power in genetic association studies. Nat Genet 37:1217–1223
De La Vega FM, Isaac H, Collins A, Scafe CR, Halldorsson BV, Su X, Lippert RA, Wang Y, Laig-Webster M, Koehler RT, Ziegle JS, Wogan LT, Stevens JF, Leinen KM, Olson SJ, Guegler KJ, You X, Xu LH, Hemken HG, Kalush F, Itakura M, Zheng Y, de The G, O’Brien SJ, Clark AG, Istrail S, Hunkapiller MW, Spier EG, Gilbert DA (2005) The linkage disequilibrium maps of three human chromosomes across four populations reflect their demographic history and a common underlying recombination pattern. Genome Res 15:454–462
The ENCODE Project Consortium (2004) The ENCODE (ENCyclopedia of DNA elements) project. Science 306:636–640
Evans DM, Cardon LR (2005) A comparison of linkage disequilibrium patterns and estimated population recombination rates across multiple populations. Am J Hum Genet 76:681–687
Eyheramendy S, Marchini J, McVean G, Myers S, Donnelly P (2007) A model-based approach to capture genetic variation for future association studies. Genome Res 17:88–95
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229
Gonzalez-Neira A, Ke X, Lao O, Calafell F, Navarro A, Comas D, Cann H, Bumpstead S, Ghori J, Hunt S, Deloukas P, Dunham I, Cardon LR, Bertranpetit J (2006) The portability of tagSNPs across populations: a worldwide survey. Genome Res 16:323–330
Gunderson KL, Kruglyak S, Graige MS, Garcia F, Kermani BG, Zhao C, Che D, Dickinson T, Wickham E, Bierle J, Doucet D, Milewski M, Yang R, Siegmund C, Haas J, Zhou L, Oliphant A, Fan JB, Barnard S, Chee MS (2004) Decoding randomly ordered DNA arrays. Genome Res 14:870–877
Huang W, He Y, Wang H, Wang Y, Liu Y, Wang Y, Chu X, Wang Y, Xu L, Shen Y, Xiong X, Li H, Wen B, Qian J, Yuan W, Zhang C, Wang Y, Jiang H, Zhao G, Chen Z, Jin L (2006) Linkage disequilibrium sharing and haplotype-tagged SNP portability between populations. Proc Natl Acad Sci USA 103:1418–1421
The International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320
Jin L, Su B (2000) Natives or immigrants: modern human origin in East Asia. Nat Rev Genet 1:126–133
Ke X, Miretti MM, Broxholme J, Hunt S, Beck S, Bentley DR, Deloukas P, Cardon LR (2005) A comparison of tagging methods and their tagging space. Hum Mol Genet 14:2757–2767
Lim J, Kim YJ, Yoon Y, Kim SO, Kang H, Park J, Han AR, Han B, Oh B, Kimm K, Yoon B, Song K (2006) Comparative study of the linkage disequilibrium of an ENCODE region, chromosome 7p15, in Korean, Japanese, and Han Chinese samples. Genomics 87:392–398
Mahasirimongkol S, Chantratita W, Promso S, Pasomsab E, Jinawath N, Jongjaroenprasert W, Lulitanond V, Krittayapoositpot P, Tongsima S, Sawanpanyalert P, Kamatani N, Nakamura Y, Sura T (2006) Similarity of the allele frequency and linkage disequilibrium pattern of single nucleotide polymorphisms in drug-related gene loci between Thai and northern east Asian populations: implications for tagging SNP selection in Thais. J Hum Genet 51:896–904
Montpetit A, Nelis M, Laflamme P, Magi R, Ke X, Remm M, Cardon L, Hudson TJ, Metspalu A (2006) An evaluation of the performance of tag SNPs derived from HapMap in a Caucasian population. PLoS Genet 2:e27
Mueller JC, Lohmussaar E, Magi R, Remm M, Bettecken T, Lichtner P, Biskup S, Illig T, Pfeufer A, Luedemann J, Schreiber S, Pramstaller P, Pichler I, Romeo G, Gaddi A, Testa A, Wichmann HE, Metspalu A, Meitinger T (2005) Linkage disequilibrium patterns and tagSNP transferability among European populations. Am J Hum Genet 76:387–398
Nejentsev S, Godfrey L, Snook H, Rance H, Nutland S, Walker NM, Lam AC, Guja C, Ionescu-Tirgoviste C, Undlien DE, Ronningen KS, Tuomilehto-Wolf E, Tuomilehto J, Newport MJ, Clayton DG, Todd JA (2004) Comparative high-resolution analysis of linkage disequilibrium and tag single nucleotide polymorphisms between populations in the vitamin D receptor gene. Hum Mol Genet 13:1633–1639
Paschou P, Mahoney MW, Javed A, Kidd JR, Pakstis AJ, Gu S, Kidd KK, Drineas P (2007) Intra- and interpopulation genotype reconstruction from tagging SNPs. Genome Res 17:96–107
Plagnol V, Wall JD (2006) Possible ancestral structure in human populations. PLoS Genet 2:e105
Ramirez-Soriano A, Lao O, Soldevila M, Calafell F, Bertranpetit J, Comas D (2005) Haplotype tagging efficiency in worldwide populations in CTLA4 gene. Genes Immun 6:646–657
Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, Lander ES (2001) Linkage disequilibrium in the human genome. Nature 411:199–204
Ribas G, Gonzalez-Neira A, Salas A, Milne RL, Vega A, Carracedo B, Gonzalez E, Barroso E, Fernandez LP, Yankilevich P, Robledo M, Carracedo A, Benitez J (2006) Evaluating HapMap SNP data transferability in a large-scale genotyping project involving 175 cancer-associated genes. Hum Genet 118:669–79
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Sawyer SL, Mukherjee N, Pakstis AJ, Feuk L, Kidd JR, Brookes AJ, Kidd KK (2005) Linkage disequilibrium patterns vary substantially among populations. Eur J Hum Genet 13:677–686
Schulze TG, Zhang K, Chen YS, Akula N, Sun F, McMahon FJ (2004) Defining haplotype blocks and tag single-nucleotide polymorphisms in the human genome. Hum Mol Genet 13:335–342
Service S, DeYoung J, Karayiorgou M, Roos JL, Pretorious H, Bedoya G, Ospina J, Ruiz-Linares A, Macedo A, Palha JA, Heutink P, Aulchenko Y, Oostra B, van Duijn C, Jarvelin MR, Varilo T, Peddle L, Rahman P, Piras G, Monne M, Murray S, Galver L, Peltonen L, Sabatti C, Collins A, Freimer N (2006) Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat Genet 38:556–560
Weale ME, Depondt C, Macdonald SJ, Smith A, Lai PS, Shorvon SD, Wood NW, Goldstein DB (2003) Selection and evaluation of tagging SNPs in the neuronal-sodium-channel gene SCN1A: Implications for linkage-disequilibrium gene mapping. Am J Hum Genet 73:551–565
Willer CJ, Scott LJ, Bonnycastle LL, Jackson AU, Chines P, Pruim R, Bark CW, Tsai YY, Pugh EW, Doheny KF, Kinnunen L, Mohlke KL, Valle TT, Bergman RN, Tuomilehto J, Collins FS, Boehnke M (2006) Tag SNP selection for Finnish individuals based on the CEPH Utah HapMap database. Genet Epidemiol 30:180–190
Yoo YK, Ke X, Hong S, Jang HY, Park K, Kim S, Ahn T, Lee YD, Song O, Rho NY, Lee MS, Lee YS, Kim J, Kim YJ, Yang JM, Song K, Kimm K, Weir B, Cardon LR, Lee JE, Hwang JJ (2006) Fine-scale map of encyclopedia of DNA elements regions in the Korean population. Genetics 174:491–497
Acknowledgments
The authors thank Sandra German at the Office of Population Studies in Cebu, the Philippines, for blood sample collection and processing, under the direction of Dr. Christopher Kuzawa of Northwest University. We thank Amy Perou of the biospecimen processing facility, Jason Luo of the mammalian genotyping core, and Laura Livingstone of the automated DNA sequencing facility at the University of North Carolina at Chapel Hill. Cebu Filipino data collection was supported by TW05596, specimen processing and genotyping was supported by pilot funds from NIH grants RR20649 (Interdisciplinary Obesity Center), ES10126 (project 7-2004-E of the Center for Environmental Health and Susceptibility), and DK56350 (Clinical Nutrition Research Center), and analysis was supported in part by grant DK78150. A.F.M. was supported by an Integrative Vascular Biology Fellowship, NIH grant HL69768.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Marvelle, A.F., Lange, L.A., Qin, L. et al. Comparison of ENCODE region SNPs between Cebu Filipino and Asian HapMap samples. J Hum Genet 52, 729–737 (2007). https://doi.org/10.1007/s10038-007-0175-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-007-0175-9
Keywords
This article is cited by
-
Comparison of genome-wide variation between Malawians and African ancestry HapMap populations
Journal of Human Genetics (2010)
-
TagSNP transferability and relative loss of variability prediction from HapMap to an admixed population
Journal of Biomedical Science (2009)
