
Abstract
We previously mapped the 41rst gene locus (DFNA41) for autosomal dominant hearing loss on chromosome 12q24-qter in a large multi-generational Chinese family. We determined that DFNA41 is located in a 15 cM region, proximal to the marker D12S1609. A maximum two point LOD score of 6.56 at θ=0.0 was obtained with marker D12S343. In the current study, screening of eight candidate genes within the DFNA41 interval did not reveal the mutation causing deafness in this family. Eight highly informative single nucleotide polymorphisms (SNPs) in the region of D12S343 were selected for linkage and association study. Because the pedigree studied here is a large family with many founders, we applied the transmission/disequilibrium (TDT) test. To account for the dependence of small families and the relatively small sample size, simulations were performed to obtain P-values. For three nearby SNPs spanning a 7 kb interval, we found significant evidence of linkage and association. The highest Z score of linkage and association of 3.6 (P≤0.0001) was obtained for SNP rs1566667. Haplotype analysis revealed that affected individuals were heterozygous for one core SNP (rs1027560–rs1027557–rs1566667–rs1463865–rs2078105) CAGTC haplotype, confirming location and autosomal dominant inheritance of the DFNA41 locus. Examination of pairwise LD calculation identified a major haplotype block defined by the four most centromeric SNPs. This study represents a significant refinement of the DFNA41 locus and should facilitate positional cloning of the disease gene.
Similar content being viewed by others

Introduction
Progressive hearing loss is a significant problem in all aging populations. It is estimated that approximately 5% of people under 45 years have a significant loss of hearing, increasing to approximately 50% by 80 years of age (Morton 1991). Age-related late onset hearing loss (presbyacusis) is considered multifactorial, with involvement of both genetic and environmental factors (Schuknecht and Gacek 1993). In contrast, childhood or adolescent hearing loss is often inherited as an autosomal dominant Mendelian trait, representing about 20% of all cases of hereditary non-syndromic sensorineural hearing impairment (NSSHI). To date, at least 53 loci for autosomal dominant NSSHI have been identified by linkage, 18 of which (with diverse function) have been cloned (see hereditary hearing loss homepage: http://dnalab-www.uia.ac.be/dnalab/hhh/). Using linkage analysis, and on the basis of recombinations in affected individuals, we previously located the DFNA41 locus in an interval defined by D12S1609 on 12q24.33 and the telomere of the q arm (Blanton et al. 2002). The candidate region of DFNA41, defined by D12S1609 and D12S357, the most distal marker in the current sequence database, spans a physical distance of about 3.9 Mb. Examination of the annotations in the web-browser interface to the genomic sequence at UCSC (http://genome.ucsc.edu/) identified more than 100 known genes and a series of predicted or poorly characterized genes in the DFNA41 candidate interval. The human chromosome region containing DFNA41 shares conserved syntheny with a segment of mouse chromosome 5, but no mutation causing hearing impairment in mice has been mapped to this region. Here, we adopted a positional and functional candidate gene strategy in an attempt to identify the DFNA41 deafness-causing gene. At the same time, we performed SNP-based linkage/association studies as an alternative approach to further establish a basis for the continuing study of candidate genes for the DFNA41 locus.
Materials and methods
Subjects
We have previously reported a six-generation Chinese family segregating for an autosomal dominant adult onset form of hearing loss (Blanton et al. 2002). Members of four generations including 36 subjects were available for testing, 22 of whom were affected (Fig. 1). Briefly, the onset of hearing loss in most of these patients was in the second decade of life, with a gradual progression from slight to severe loss over several decades. There were no obvious vestibular dysfunction or other associated abnormalities. In all affected individuals, audiometry showed a bilateral and symmetrical sensorineural hearing loss involving all frequencies.

Pedigree and haplotype analysis of the Chinese family that segregates DFNA41. Black symbols Affected subjects. Haplotypes are represented by bars, with the haplotype associated with hearing loss in black
Screening of candidate genes
Genes located in the candidate interval that are enriched in the inner ear and that contain domains that suggest cochlea-related functions, were screened for mutation by searching the Inner Ear Gene Expression Database (http://www.mgh.harvard.edu/depts/coreylab/index.html). These genes include Zinc finger protein 10 (ZFOC1) (AL834266; Unigene cluster, Hs.61976), Q8N3T6 (XM_044062; Hs.49599), AK056047 (gi: 16551125; Hs.199630), Frizzled-10 (FZD10) (NM_007197; Hs.31664), RIM binding protein 2 (KIAA0318; NM_015347; Hs.225014), Epimorphin (EPIM; BC0474496; Hs.99865), Q86SM4 (AY255587; Hs.158258), KIAA0692 (XM_208619, Hs.289019). The positions of the candidate genes on the long arm of the chromosome 12 are based on maps available from the UCSC genome browser (human July assembly; http://genome.ucsc.edu/). The sequences of coding regions and intron–exon junctions of the genes were determined using either the web-browser interface to the genomic sequence at UCSC or Ensembl (http://www.ensembl.org/). Primers were designed for polymerase chain reaction (PCR) amplification of coding regions and exon–intron boundaries of the candidate genes. The resulting PCR products were gel-purified and sequenced using an ABI PRISM BigDye Terminator Cycle Sequencing reaction kit (Applied Biosystems, Foster City, CA). An automated sequencer (ABI 3100) was used for direct sequencing. Genomic DNA obtained from blood lymphocytes from affected and unaffected members of the family was sequenced.
Single nucleotide polymorphism markers
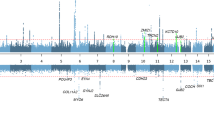
Eight single nucleotide polymorphisms (SNPs) in the region of D12S343—a marker for which a maximum 2 point lod score of 6.56 at θ=0.0 was obtained—were used for association study (Fig. 2). SNPs rs1566668, rs1566669, rs1027560, rs1027557, rs156667, rs937533, rs1463865, rs2078105, spanning a distance of ∼60.8 kb, were selected from the SNP consortium (TSC) database as having a minor allele frequency of at least 20% in Asian populations (http//www.snp.cshl.org). The base positions of the SNPs were determined from maps at the UCSC genome browser (human July assembly; http://genome.ucsc.edu/). A combination of single-strand conformation polymorphism (SSCP) and direct sequencing methods was used for genotyping. For SSCP analysis, the PCR products were initially run on a 1 mm-thick 8% non-denaturing polyacrylamide gel (acrylamide: N, N′-methylene: bisacrylamide 49:1) at 4°C. SSCPs were subsequently detected using silver staining as previously described (Liu et al. 1997).

Schematic physical map of the 12q24.32-qter, showing the markers used in the present study. The genes analyzed are indicated. The marker order and intermarker distances are based on maps available at UCSC (http://genome.ucsc.edu/). Deafness-associated single nucleotide polymorphisms (SNPs) are shown in bold. For clarity, representation of physical distances is not to scale
Linkage and association studies
We adopted a within-family Monte Carlo procedure, performed by a computer program to test for linkage and association of SNP to disease. The Z score for each of the SNPs was calculated using the program Family-Based Association Test (FBAT; Martin et al. 2000, 2001). FBAT breaks down the large family into several small families. To account for the dependence of small families and the relatively small sample size, we performed simulations to obtain P-values. To test linkage, we first retained the family structure and all the founder’s genotypes. The genotypes of individuals whose parents have not been genotyped were also retained. The genotypes of the remaining individuals in fourth, fifth, and sixth generations were randomly assigned according to their parental genotypes by assuming random transmission. This process was repeated 10,000 times and each time a Z score was calculated using FBAT. The empirical P-value of testing for linkage is assigned by noting the proportion of replicates in which this number is equal to or more extreme than the observed value in the actual data. To test for association, we first simulated all the founder’s genotypes according to the allele frequencies. Offspring genotypes were assigned assuming random transmission from parents. The genotypes of the individuals whose original genotypes were missing were again assigned the value missing. The process was repeated 10,000 times and Z scores for each replication were calculated using FBAT. The empirical P-value was obtained similarly as for linkage testing. Finally, we inferred haplotypes for each individual using SIMWALK2 (Sobel and Lange 1996), and an association test was similarly performed. The pairwise LD parameter Lewontin’s D′ (Lewontin 1964) was calculated based on unrelated haplotypes extracted from the whole family.
Results
Screening of candidate genes
Eight genes in the DFNA41 candidate interval were selected based on the potential for their involvement in the pathophysiology of hearing, and based on their presence in the Inner Ear Gene Expression Database (http://www.mgh.harvard.edu/depts/coreylab/index.html) (Table 1). We first screened for mutations in an obvious candidate gene, FZD10, a member of the frizzled gene family. DNA sequencing of coding and exon–intron boundaries of the FZD10 gene in DFNA41-affected subjects did not reveal a disease causing mutation. However, one known common variation located in the promoter region at position −139, resulting in a G to A substitution (rs1875121) was detected, but no association between the development of the deafness phenotype and this sequence change was found. At present, it is not known whether this polymorphism has any effect on the protein.
We next tested another promising candidate gene, KIAA0318 encoding RIM binding protein 2, which contains three SH3 domains and three fibronectin type III motifs. These motifs are most commonly observed in proteins comprising components of the basal lamina and extracellular matrices, and in cell adhesion molecules. This gene could be excluded as a DFNA41-causative gene, as no disease-causing allele variant was found by sequencing the coding sequences and exon–intron boundaries. However, a total of 11 polymorphisms were detected in the KIAA0318 gene, 4 of which have been previously reported. Seven of the polymorphisms are located in coding regions. Of these, only 853G>C (A285P) in exon 7 leads to a non-synonymous substitution in the translated protein. Four of the polymorphisms occur within intronic regions (Table 1).
Another strong candidate gene tested in the present study was epimorphin. The epimorphin protein regulates epithelial–mesenchymal interactions, and epithelial cell morphogenesis and activation. Sequence analysis of human epimorphin in the family revealed only two polymorphisms, including a previously reported non-synonymous change due to a G→C transversion (rs6486602) that is predicted to cause a Ser42Thr amino acid substitution. The second polymorphism found in the epimorphin gene involves an A→G change at a position located within intron 9.
Q86SM4 (also known as PGR25) was another candidate gene tested. The gene product, which has seven hydrophobic transmembrane domains, is believed to interact with G-proteins. The PGR25 protein would have a G-protein-coupled receptor activity and might be involved in cellular trafficking and/or be implicated in neuropeptide signaling pathways. However, sequence analysis of the PGR25 gene in the DFNA41 family revealed only two intronic changes in the gene: IVS3-34G>A and IVS4+61T>C.
Two genes with no documented function, but which are within the candidate interval and enriched in the inner ear, were also investigated, but no deafness-related sequence change was found. However, synonymous polymorphisms, 468A→G, 492A→G in exon 3 and 1005 A→G in exon 4, as well as the INS3-44A→G intronic change in Q8N3T6 were detected. Likewise, in AK056047, two non-synonymous polymorphisms, 2122T→G and 2392C→A, which result in X710G and L798I substitutions, respectively, and a deletion of a G followed by three base changes replacing TTT by CCC in intron 4 were detected, which were also present in several control individuals with normal hearing. Finally, sequencing of the exons and flanking regions of ZFOC1 (encoding a zinc finger protein) and of KIAA0692 (encoding a protein containing an ankyrin repeat) also revealed no variation (Table 1).
Linkage and association study
The absence of recombination between the DFNA41 locus and D12S343 does not infer that all the SNPs in the region are in complete linkage disequilibrium (LD) with the disease variant. Because of the presence of several founders in our multi-generational family, we were able to perform a linkage/association study to compare the allele frequencies between affected and unaffected individuals. A within-family Monte Carlo permutation procedure was applied to test linkage/association of an SNP to the deafness phenotype, using FBAT. A panel of eight SNPs covering ∼60.8 kb was selected from the DFNA41-linked region (proximal to D12S343) for the present study (Fig. 2).
Three markers showed significant linkage and association with the deafness phenotype, indicating the existence of LD with the disease locus over the region investigated (Table 2).
There was an overtransmission of allele G of SNP 1566667 and allele A of SNP 1027557 to the affected subjects, with Z=3.63 (P≤0.0001 for linkage or association based on 10,000 replicates) and Z=3.528 (P≤0.0001 for linkage or association based on 10,000 replicates), respectively. Lower, but still very significant, association was observed with the C allele of SNP 1027560, located downstream (centromeric), with Z=2.5 (P=0.0085 for linkage and 0.0172 for association). The three SNPs cover a distance of ∼7 kb. Similar tests for SNPs 2078105, 1463865, 937533, 1566669, 1566668 were not significant. Haplotype analysis revealed that haplotype CAGTC (rs1027560, rs1027557, rs1566667, rs1463865, rs2078105) is also significantly associated with the deafness phenotype (P=0.0187). This haplotype appears in all affected individuals in a heterozygous state, supporting autosomal dominant inheritance of the DFNA41 locus (Fig. 1). We next defined the pattern of LD to ensure that we had adequate SNP coverage for our association study, and achieved sufficient SNP spacing for an association examination to be performed. Seven of the SNPs shown in Fig. 2, except SNP rs1566669, which was not informative for the family, were used for analysis. Examination of pairwise LD measured by Lewontin’s D′ suggests one major haplotype block consisting of four SNPs (rs1566668, rs1027560, rs1027557 and rs1566667) with a somewhat weak level of LD among SNPs rs937533, rs1463865 and rs2078105 (Table 3). Our data are consistent with significant allelic association of SNPs rs1027560, rs1027557 and rs1566667 to deafness.
Discussion
Our previous genome-wide scan had shown linkage to the DFNA41 locus in an interval defined by D12S1609 on 12q24.33 and the telomere of the long arm of chromosome 12 (Blanton et al. 2002). Another dominant gene for deafness, DFNA25, from a large family of Czech descent in the United States has been mapped to 12q21–24 between D12S327 and D12S84 (Greene et al. 2001). By a multi-point analysis, we clearly excluded the DFNA25 locus and placed the DFNA41 locus at 46 cM telomeric to D12S84.
The LD mapping method is a powerful tool for genetic mapping in isolated founder populations. Several disease-related genes have been mapped and cloned using this method, such as those responsible for diastrophic dysplasia, progressive myoclonus epilepsy, and Fukuyama muscular dystrophy, in Finnish and Japanese populations (Hastbacka et al. 1992; Jorde 1995; Toda et al. 1996). Because the pedigree we studied here is a large multi-generational family with many founders, we were able to apply the transmission/disequilibrium (TDT) test. To account for the dependence of small families and the relatively small sample size, we conducted a simulation approach by generating the data under the null hypothesis of no linkage or association, which is more robust than using asymptotical distribution as the test statistic. Since the permutation is carried out within families, potential problems resulting from population structure are eliminated. Of the eight SNPs (spaced at 0.2–35 kb intervals) in the ∼60 kb proximal to D12S343 used in the present study, three SNPs within a 7 kb interval showed association with deafness. LD is known to vary markedly over different chromosomal regions and distances. In addition, average LD measures cannot be accurately predicted from one chromosomal region to another (Abecasis et al. 2001; Reich et al. 2001; Stephens et al. 2001) and LD is structured into “blocks” of DNA that can range from ∼5 to 100 kb (Daly et al. 2001; Jeffreys 2001). In the present study, we observed a major block of LD delimited by the four most centromeric SNPs, and LD was substantially weaker with the other three SNPs within the candidate region of DFNA41. The results are consistent with the findings from allelic association analysis of the three SNPs in the block to deafness. The results also suggest that the disease “susceptibility” allele may be located within the block. However, some limitations of this study should be acknowledged, and there are reasons to be cautious about interpretation of data due to the small sample size. Only a limited number of independent haplotypes within the extended pedigrees were available for calculations of the LD between the SNPs. However, our simulation data suggested that SNP1566667 as well as SNP1027557 are significantly associated with deafness, even after the correction for multiple tests.
Association is the non-random cosegregation of alleles and assumes that populations are descended from a small founder group and that repeated recombinations over generations reduce the shared chromosomal segments to very small regions. Therefore, in order to detect an association, the marker and disease gene must be in LD. Because LD extends to shorter distances, demonstration of association refines the region likely to harbor the disease gene. Association could arise due to LD with a disease mutation in a nearby gene and thus, in the present study, may imply a close physical linkage of the three deafness-associated SNPs within a 7 kb interval and the disease gene. Thus, our current linkage/association studies provide a basis for the continuing study of candidate genes for the DFNA41 locus. In this respect, the three disease-associated SNPs that are located within the candidate interval may act as pointers to identify which genomic regions to analyze. Alternatively, these disease-associated SNPs may indicate locus control elements, either for genes within the region or for genes outside the critical region. None of the three disease-associated SNPs are located within the protein-coding or putative promoter regions of the tested candidate genes. However, the most distal SNP used in the present study (SNP2078105; Fig. 2) that is not associated with the deafness phenotype, is located ∼8 kb centromeric (downstream) of the 5′-end of the Frizzled-10 (FZD10) gene. The most telomeric disease-associated SNP 1566667 lies 23 kb downstream of the 3′-end of the AK056047 gene.
The dominant inheritance of the DFNA41 allele seemed most consistent with a missense coding mutation; however, dominance can also result from other mechanisms such as inactivation of candidate genes, regulatory effects, null alleles and/or segmental deletions (haploinsufficiency), or segmental duplications. The possibility of inversions or other gross rearrangements should also be considered. We screened eight potential inner ear enriched genes for mutation but DNA sequencing of coding and exon–intron boundaries of the genes in affected subjects did not reveal any disease-causing mutation. However, there are several types of mutations that DNA sequencing alone would be unable to detect. These include any deletion or duplication (resulting in hemizygosity or gene amplification, respectively) with breakpoints outside of the relatively small coding elements that we sequenced. The association results presented here pointed to a candidate region defined by the SNPs 1566667 and 1027560, which should be our primary “hot-spot” for the location of the DFNA41 gene on 12q24.33. The ultimate cloning of the gene causing DFNA 41 will provide further insights into our understanding of the molecular pathophysiology of late-onset hearing loss.
References
Abecasis GR, Noguchi E, Heinzmann A, Traherne JA, Bhattacharyya S, Leaves NI, Anderson GG, Zhang Y, Lench NJ, Carey A, Cardon LR, Moffatt MF, Cookson WO (2001) Extent and distribution of linkage disequilibrium in three genomic regions. Am J Hum Genet 68:191–197
Blanton SH, Liang CY, Cai MW, Pandya A, Du LL, Landa B, Mummalanni S, Li KS, Chen ZY, Qin XN, Liu YF, Balkany T, Nance WE, Liu XZ (2002) A novel locus for autosomal dominant non-syndromic deafness (DFNA41) maps to chromosome 12q24-qter. J Med Genet 39:567–570
Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES (2001) High-resolution haplotype structure in the human genome. Nat Genet 29:229–232
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229
Greene CC, McMillan PM, Barker SE, Kurnool P, Lomax MI, Burmeister M, Lesperance MM (2001) DFNA25, a novel locus for dominant nonsyndromic hereditary hearing impairment, maps to 12q21-24. Am J Hum Genet 68:254–260
Hastbacka J, de la Chapelle A, Kaitila I, Sistonen P, Weaver A, Lander E (1992) Linkage disequilibrium mapping in isolated founder populations: diastrophic dysplasia in Finland. Nat Genet 2:204–211
Jeffreys AJ, Kauppi L, Neumann R (2001) Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet 29:217–222
Jorde LB (1995) Linkage disequilibrium as a gene-mapping tool. Am J Hum Genet 56:11–14
Kruglyak L (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 22:139–144
Lewontin RC (1964) The interaction of selection and linkage I. General considerations; heterotic models. Genetics 49:49–67
Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, Brown SD (1997) Mutations in the myosin VII a gene cause nonsyndromic recessive deafness. Nat Genet 16:188–190
Martin ER, Monks SA, Warren LL, Kaplan NL (2000) A test for linkage and association in general pedigrees: the pedigree disequilibrium test. Am J Hum Genet 67:146–154
Martin ER, Bass MP, Kaplan NL (2001) Correcting for a potential bias in the pedigree disequilibrium test. Am J Hum Genet 68:1065–1067
Morton NE (1991) Genetic epidemiology of hearing impairment. Ann NY Acad Sci 630:16–31
Nickerson DA, Taylor SL, Weiss KM, Clark AG, Hutchinson RG, Stengard J, Salomaa V, Vartiainen E, Boerwinkle E, Sing CF (1998) DNA sequence diversity in a 9.7-kb region of the human lipoprotein lipase gene. Nat Genet 19:233–240
Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, Lander ES (2001) Linkage disequilibrium in the human genome. Nature 411:199–204
Schuknecht HF, Gacek MR (1993) Cochlear pathology in presbycusis. Otol Rhinol Laryngol 102:1–116
Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 58:1323–1337
Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, Jiang R, Messer CJ, Chew A, Han JH, Duan J, Carr JL, Lee MS, Koshy B, Kumar AM, Zhang G, Newell WR, Windemuth A, Xu C, Kalbfleisch TS, Shaner SL, Arnold K, Schulz V, Drysdale CM, Nandabalan K, Judson RS, Ruano G, Vovis GF (2001) Haplotype variation and linkage disequilibrium in 313 human genes. Science 293:489–493
Toda T, Miyake M, Kobayashi K, Mizuno M, Saito K, Osawa M, Nakamura Y, Kanazawa I, Nakagome Y, Tokunaga K, Nakahori Y (1996) Linkage disequilibrium mapping narrows the Fukayama-type congenital muscular dystrophy (FCMD) candidate region to 100 kb. Am J Hum Genet 59:1313–1320
Acknowledgements
The authors would like to thank the family members for participating in this study. This research is supported by NIH grant DC 05575 (to X.Z.L.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yan, D., Ouyang, X.M., Zhu, X. et al. Refinement of the DFNA41 locus and candidate genes analysis. J Hum Genet 50, 516–522 (2005). https://doi.org/10.1007/s10038-005-0286-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-005-0286-0
Keywords
This article is cited by
-
Determining the effectiveness of High Resolution Melting analysis for SNP genotyping and mutation scanning at the TP53 locus
BMC Genetics (2009)
-
The genetic bases for non-syndromic hearing loss among Chinese
Journal of Human Genetics (2009)
