
Abstract
Heat shock protein 27 (HSP27) belongs to a family of small heat shock proteins that play significant roles in the cellular stress response and are also involved in the control of protein–protein interactions as chaperons. Mutation in HSP27 has been identified as the cause of axonal Charcot–Marie–Tooth disease (CMT) and distal hereditary motor neuropathy (HMN). Heat shock protein 22 (HSP22) is a molecular counterpart of HSP27, and its mutation is another cause of distal HMN. We screened the mutation of HSP27 and HSP22 in 68 Japanese patients with axonal CMT or unclassified CMT and six Japanese patients with distal HMN. We detected a heterozygous P182S mutation of HSP27 in a patient with distal HMN, but we found no mutations in HSP22. Mutation in HSP27 may impair the formation of the stable neurofilament network that is indispensable for the maintenance of peripheral nerves.
Similar content being viewed by others

Introduction
Charcot–Marie–Tooth disease (CMT) is one of the most common but heterogeneously hereditary motor and sensory neuropathy. CMT has been classified into two types: CMT1 and CMT2, the demyelinating and axonal forms, respectively. CMT2 has been further divided into nine subgroups by linkage studies (Takashima et al. 1999; Mersiyanova et al. 2000; Ismailov et al. 2001; Antonellis et al. 2003; Klein et al. 2003; Verhoeven et al. 2003; Nelis et al. 2004; Tang et al. 2004; Zuchner et al. 2004). Distal hereditary motor neuropathy (HMN) is an exclusively motor neuropathy but is also a clinically and genetically heterogeneous neuropathy. Distal HMN cannot be clearly distinguished from CMT2 because sensory signs are often lacking in CMT2. Evgrafov et al. (2004) detected the mutations of the heat shock protein 27 (HSP27) gene in a Russian family with CMT2F and in patients with CMT2 or distal HMN. HSP27 belongs to a large protein family of the small heat shock proteins. Heat shock protein 22 (HSP22) is also another member of the small heat shock proteins, and its missense mutations have been detected in two families with distal HMN (Irobi et al. 2004). The small heat shock proteins play a marked role in cellular stress response. They are implicated in many different cellular processes, such as suppression of protein aggregation and involvement in the dynamics of cytoskeletal proteins, cellular growth, transcription, and differentiation (Perng et al. 1999; Benn et al. 2002; Gabai and Sherman 2002; Vleminckx et al. 2002).
In the present report, we studied HSP27 and HSP22 in 74 Japanese patients with axonal CMT, including those with unclassified CMT or distal HMN, and detected a mutation of HSP27 in one patient with distal HMN.
Subjects and methods
Subjects
We studied 59 axonal CMT patients [male-to-female ratio 38:21; age at analysis 35.7±23.0 (mean ± SD) years; age at onset 21.8±20.4 years), nine unclassified CMT patients (male-to-female ratio 6:3; age at analysis 30.0±16.4 years; age at onset 19.0±16.9 years), and six distal HMN patients (male-to-female ratio 3:3; age at analysis 22.5±18.4 years; age at onset 12.8±9.7 years). All these patients did not have any mutation of Po and Cx32. CMT2 and distal HMN were diagnosed according to criteria recommended by the European CMT Consortium (De Jonghe et al. 1998). Axonal CMT patients included 22 familial (12 dominant and ten probably recessive) and 37 isolated cases showing CMT2 phenotype. Unclassified CMT patients included five familial (one dominant and four probably recessive) and four isolated cases. All distal HMN patients were isolated cases. HSP27 mutation was identified in the following patient with distal HMN.
Case 251
The patient was the second child of unrelated, healthy parents. No family members had similar symptoms. He had asphyxia at birth and hyperbilirubinemia during the neonatal period, but he developed normally. He had fallen frequently from age four and had a right drop foot and leg pain at walking from age eight. On examination at age 15, he presented steppage gait, pes cavus, drop feet, and distal muscle weakness and atrophy of the lower extremities but had no sensory disturbances and no decreased deep-tendon reflexes. He had normal motor (64.2 m/s) and sensory (51.8 m/s) nerve conduction velocities of right median nerves and normal amplitude (30 μV) of sensory action potential of right median nerve. A sural nerve specimen for histological analysis was not taken.
Gene analyses
The Ethics Committee of the Yamagata University School of Medicine approved this study. With written informed consent from the patients and their families, peripheral blood specimens were used for genomic DNA extraction. DNA of healthy controls was also prepared from Japanese medical students and coworkers who agreed to the study protocol. All coding exons including exon–intron boundaries of HSP27 (NP_001531) and HSP22 (NP_055180) were amplified by the polymerase chain reaction (PCR) with the primers designed according to the data of Homo sapiens chromosome 7 (NC_000007) and 12 (NT_009775) genomic contig, respectively. We screened the mutation by denaturing high-performance liquid chromatography analysis (DHPLC) (Transgenomic WAVE system). The fragments showing heteroduplex were sequenced by the Dye Deoxy Terminator Cycle method on an ABI PRISM Genetic Analyzer 310 (PE Applied Biosystems, Foster City, CA, USA).
Paternity confirmation
Paternity was confirmed using 16 markers provided with an AmpFLSTR identifier Kit (PE Applied Biosystems).
Results
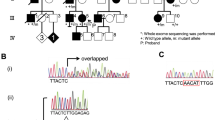
By screening all coding regions of HSP27 and HSP22 using DHPLC, we detected DNA fragments showing heteroduplex and determined these sequences. Sequencing analysis of the fragment revealed that a patient with distal HMN was heterozygous for P182S of HSP27 (Fig. 1). The P182S mutation was not detected in his parents or his elder brother. His paternity was confirmed using polymorphic markers, as described in Human subjects and methods, indicating that the P182S mutation was a de novo mutation. The P182S mutation was also not detected in the 100 normal chromosomes and was predicted to change an amino acid conserved in the orthologs of other species, such as the mouse (NP_038588), rat (NP_114176), Canis familiaris (P42929), Cricetulus longicaudatus (CAA36036), Gallus gallus (A49181), and Poeciliopsis lucida (O13227). We also detected several polymorphic nucleotide substitutions as follows; c.1-56C>T, c.365-7C>T and c.108C>G (P36P).

Sequence chromatography of the family. Sequence chromatography of the patient revealed a heterozygous C-to-T mutation at nucleotide 544 (arrow) in HSP27, resulting in a P182S substitution.
As for HSP22, we found no mutations except for polymorphic mutations such as c.24C>T (F8F), c.431+83C>T, c.431+135A>C and c.582C>T (T194T).
Discussion
We studied HSP27 and HSP22 in 74 patients with axonal CMT, including those with unclassified CMT or distal HMN, and detected the P182S mutation of HSP27 in a patient with distal HMN.
The heat shock proteins belong to a large group of peptides, the stress proteins, which are induced in response to environmental challenges and developmental transitions. Small heat shock proteins are also involved in the control of protein–protein interactions as chaperons. They interact with intermediate filaments and prevent their aggregation (Perng et al. 1999). Neurofilament, a neuron-specific, intermediate protein that consists of neurofilament light (NEFL), neurofilament medium, and neurofilament heavy, plays a significant role in the maintenance of the axonal cytoskeleton and transport. Mutations in NEFL cause axonal CMT (CMT2E) and are predicted to alter the formation of the normal, intermediate filament network (Mersiyanova et al. 2000). Upregulation of HSP27 was observed in several neurodegenerative diseases and is required for the survival of injured sensory and motor neurons (Benn et al. 2002). Mutation in HSP27 may inhibit the formation of the neurofilament network and impair the maintenance of the axonal cytoskeleton and transport (Evgrafov et al. 2004).
Five mutations of HSP27 in the five patients with axonal CMT or with distal HMN have been reported so far. Four of them were located in the Hsp20-alpha-crystallin domain, a highly conserved domain among the human heat shock proteins (Evgrafov et al. 2004). The other, the P182L mutation, occurred in the variable C-terminal tail of the protein (Evgrafov et al. 2004). The P182 residue is close to dominant negative mutations of alphaB -crystallin that cause myofibrillar myopathy and affect chaperone function (Vicart et al. 1998; Selcen and Engel 2003). P182 is conserved in the orthologs of many species, and the P182S mutation is predicted to change the secondary structure. Together with the absence of P182S mutation in 100 normal chromosomes and the de novo mutation in a sporadic case, P182S mutation is likely a disease-causing mutation. The P182S mutation may act in a dominant negative manner as C-terminal mutations of alphaB-crystallin.
Mutation in HSP22 is the cause of distal HMN. Considering HSP22 is a molecular counterpart of HSP27, we also studied HSP22 in patients with axonal CMT, including those with unclassified CMT as candidates (Irobi et al. 2004). However, we could not find any mutations of HSP22 in the patients with axonal CMT. Recently, Tang et al. (2004) analyzed HSP22 in 114 patients with CMT2 and found a mutation in one family.
Mutations of HSP22 and HSP27 are not prevalent but are definite causes of axonal CMT and distal HMN.
References
Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, (2003) Glycyl tRNA synthetase mutations in Charcot–Marie–Tooth disease type 2D and distal spinal muscular atrophy type V Am J Hum Genet. 72:1293–1299
Benn SC, Perrelet D, Kato AC, Scholz J, Decosterd I, Mannion RJ, (2002) Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival Neuron 36:45–56
De Jonghe P, Timmerman V, Van Broeckhoven C, workshop participants (1998) Second workshop of the European CMT consortium: 53rd ENMC international workshop on classification and diagnostic guidelines for Charcot–Marie–Tooth type 2 (CMT2-HMSN II) and distal hereditary motor neuropathy (distal HMN-spinal CMT) 26–28 September 1997, Naarden, The Netherlands. Neuromuscul Disord 8:426–431
Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, (2004) Mutant small heat-shock protein 27 causes axonal Charcot–Marie–Tooth disease and distal hereditary motor neuropathy Nat Genet. 36:602–606
Gabai VL, Sherman MY, (2002) Invited review: interplay between molecular chaperones and signaling pathways in survival of heat shock J Appl Physiol 92:1743–1748
Irobi J, Van Impe K, Seeman P, Jordanova A, Dierick I, Verpoorten N, (2004) Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy Nat Genet 36:597–601
Ismailov SM, Fedotov VP, Dadali EL, Polyakov AV, Van Broeckhoven C, Ivanov VI, (2001) A new locus for autosomal dominant Charcot–Marie–Tooth disease type 2 (CMT2F) maps to chromosome 7q11-q21 Eur J Hum Genet 9:646–650
Klein CJ, Cunningham JM, Atkinson EJ, Schaid DJ, Hebbring SJ, Anderson SA, (2003) The gene for HMSN2C maps to 12q23–24: a region of neuromuscular disorders Neurology 60:1151–1156
Mersiyanova IV, Perepelov AV, Polyakov AV, Sitnikov VF, Dadali EL, Oparin RB, (2000) A new variant of Charcot–Marie–Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene Am J Hum Genet. 67:37–46
Nelis E, Berciano J, Verpoorten N, Coen K, Dierick I, Van Gerwen V, (2004) Autosomal dominant axonal Charcot–Marie–Tooth disease type 2 (CMT2G) maps to chromosome 12q12-q13.3 J Med Genet. 41:193–197
Perng MD, Cairns L, van den IJssel P, Prescott A, Hutcheson AM, Quinlan RA, (1999) Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin J Cell Sci. 112:2099–2112
Selcen D, Engel AG, (2003) Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations Ann Neurol 54:804–810
Takashima H, Nakagawa M, Suehara M, Saito M, Saito A, Kanzato N, (1999) Gene for hereditary motor and sensory neuropathy (proximal dominant form) mapped to 3q13.1 Neuromuscul Disord 9:368–371
Tang BS, Luo W, Xia K, Xiao JF, Jiang H, Shen L, (2004) A new locus for autosomal dominant Charcot–Marie–Tooth disease type 2 (CMT2L) maps to chromosome 12q24 Hum Genet 114:527–533
Tang BS, Zhao GH, Luo W, Xia K, Cai F, Pan Q, (2005) Small heat-shock protein 22 mutated in autosomal dominant Charcot–Marie–Tooth disease type 2L Hum Genet. 116:222–224
Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer-Grumbach M, Kwon JM, (2003) Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot–Marie–Tooth type 2B neuropathy Am J Hum Genet. 72:722–727
Vicart P, Caron A, Guicheney P, Li Z, Prevost MC, Faure A, (1998) A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy Nat Genet. 20:92–95
Vleminckx V, Van Damme P, Goffin K, Delye H, Van Den Bosch L, Robberecht W, (2002) Upregulation of HSP27 in a transgenic model of ALS J Neuropathol Exp Neurol. 61:968–974
Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot–Marie–Tooth neuropathy type 2A Nat Genet. 36:449–504
Acknowledgements
This work was supported by a Grant-in-Aid for COE Research and a Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Science, Culture and Sports of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kijima, K., Numakura, C., Goto, T. et al. Small heat shock protein 27 mutation in a Japanese patient with distal hereditary motor neuropathy. J Hum Genet 50, 473–476 (2005). https://doi.org/10.1007/s10038-005-0280-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-005-0280-6
Keywords
This article is cited by
-
Small heat shock proteins in neurodegenerative diseases
Cell Stress and Chaperones (2020)
-
Mutations in HspB1 and hereditary neuropathies
Cell Stress and Chaperones (2020)
-
Controlling properties of human neural progenitor cells using 2D and 3D conductive polymer scaffolds
Scientific Reports (2019)
-
Mutant HSPB1 causes loss of translational repression by binding to PCBP1, an RNA binding protein with a possible role in neurodegenerative disease
Acta Neuropathologica Communications (2017)
-
Proline isomerization in the C-terminal region of HSP27
Cell Stress and Chaperones (2017)
