
Abstract
Y-chromosomes from 76 Chinese men covering 33 ethnical minorities throughout China as well as the Han majority were collected as genetic material for the study of Chinese nonrecombinant Y-chromosome (NRY) phylogeny. Of the accepted worldwide NRY haplogroups, three (haplogroups D, C, O) were significant in this sample, extending previous assessments of Chinese genetic diversity. Based on geographic, linguistic, and ethnohistorical information, the 33 Chinese ethnical minorities in our survey were divided into the following four subgroups: North, Tibet, West, and South. Inferred from the distribution of the newfound immediate ancestor lineage haplogroup O*, which has M214 but not M175, we argue that the southern origin scenario of this most common Chinese Y haplogroup is not very likely. We tentatively propose a West/North-origin hypothesis, suggesting that haplogroup O originated in West/North China and mainly evolved in China and thence spread further throughout eastern Eurasia. The nested cladistic analysis revealed in detail a multilayered, multidirectional, and continuous history of ethnic admixture that has shaped the contemporary Chinese population. Our results give some new clues to the evolution and migration of the Chinese population and its subsequence moving about in this land, which are in accordance with the historical records.
Similar content being viewed by others
Introduction
Binary polymorphisms on the nonrecombining portion of the Y-chromosome (NRY) have recently been widely used to trace the origin and migration events of modern paternal lineages. Paternally transmitted and free of recombination, NRY markers can be used to construct an unequivocal and exclusive phylogeny of the Y-chromosome in the global population. Studies of the geographical and ethnical structure of phylogeny lead to the well-known “out-of-Africa” hypothesis of modern human evolution and the subsequent peopling of the entire world. New binary markers allow for the construction of more robust genetic phylogenies than before (Underhill et al. 1996; Hammer et al. 1998; Karafet et al. 1999; Su et al. 1999, 2000; Thomson et al. 2000; Underhill et al. 2000; Hammer et al. 2001; Karafet et al. 2001; Underhill et al. 2001; Wells et al. 2001; Lell et al. 2002; Qamar et al. 2002; Tajima et al. 2002; Zerjal et al. 2002).
The history of the Chinese 5,000-year civilization is a history of diverse ethnical admixture. The genetic structure of the Chinese population is a rich contemporary paternal gene pool that helped to further uncover the trails of the eastern Asian population history and the ongoing demographic processes. However, previous studies had relatively few Chinese subjects, and little information on intra-Chinese events can be deduced from them (Su et al. 1999, 2000). Because of its size and historical importance, China has figured prominently in attempts to reconstruct the human colonization of Earth (Davis and Ranov 1999; Goebel 1999). China has been placed at the confluence of northern and southern routes in the original human expansion (Underhill et al. 2001). Further, recent work has assumed a common ancestry of north and south Chinese with genetic differences between them attributed to secondary isolation by distance (IBD) mitigated by periodic interbreeding (Ding et al. 2000; Yao et al. 2002). Su et al. (1999) proposed the southern origin scenario with genetic evidence using the M214/M175 lineage.
An understanding of the genetic diversity of modern Chinese cannot ignore the distinct genomes of non-Han Chinese. According to government statistics, 93% of Chinese belong to the ethnic majority Han, and the additional 55 officially recognized minorities together account for the remaining 7% (see Cavalli-Sforza 1998 for a review). In the historical past, however, the Han were less dominant, and many of the ethnic minorities were more numerically significant. Some ethnic minorities are believed to have played important roles in East Asian migration waves: Hun and Turks have successively pastured in North China; Mongolian and Manchu, coming from the north and northeast of China, have dominated China empires; Di-Qiang had an important active role as nomads in the west of China; Yi and Bai were once rulers of southwestern China; Miao, originating with Han from the Central Plain, dispersed to the whole of South China (Li 1987; Ge et al. 1997; Fei 1999).
For this study, Chinese ethnic minorities were the focus. All samples were assayed for new NRY binary markers as well as previously published ones.
Materials and methods
Subjects
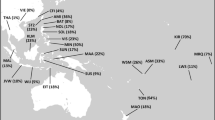
Seventy-six new blood samples of young men covering 34 Chinese populations (33 ethnic minorities and Han) from throughout China were collected as genetic material for this study. Populations sampled were selected based on their numerical significance and ethnologically postulated distinctive migration history. Families of the sampled males had lived at their present locations for at least three generations. All samples were collected with appropriate ethical approval and informed consent. The 33 Chinese ethnic minorities were divided into the following four subgroups based on geographic, linguistic, and ethnohistorical data. Detailed classification information about main geographical distributions and linguistic families of Chinese populations surveyed is shown in Table 1, and the approximate geographic locations of the study samples are shown in Fig. 3.
The “North” subgroup included Mongolian, Daur, Manchu, Hezhe, Xibo, Korean, and Yugur. These populations inhabit North China and are members of the Altaic linguistic family (Wei and Wang 2000). Tibetan, Yi, Naxi, Lahu, Hani, Qiang, Primi, Bai, Nu, and Derung were combined into the “Tibet” subgroup. These populations live in the plateau of Southwest China and are members of the Tibeto-Burman linguistic family, a subgroup of the Sino-Tibetan linguistic family (Wei and Wang 2000). The “West” subgroup is comprised of Uyghur, Kazak, Kirghiz, Hui, Salar, and Dongxiang. Although their individual histories and linguistic families vary, they are all Muslim minorities in China, and the origins of the six populations have been traced to Arab, Iranian, and Central Asian sources, and/or the Mongolian peoples. Except Hui, their current main geographical distributions are all in West China (Wong and Dajani 1988; Gladney 1996; Wei and Wang 2000). Miao, Yao, She, Zhuang, Dong, Bouyei, Shui, Gelao, Tujia, and Va were joined as the “South” subgroup. These populations occupy the vast territory of South China and are members of the Sino-Tibetan linguistic family except for the Va (Austro-Asiatic linguistic family) (Wei and Wang 2000).
Binary markers and phylogeny construction
NRY SNPs that varied within our sample were used as binary markers. A 19-kb fragment of the SMCY gene (Genbank accession: AF273841, between 31,885 and 50,983 bp) was selected to be sequenced in steps. First, three 8-kb contiguous from the 19-kb fragment were amplified using long template accurate (LA) PCR. The resulting DNA, diluted 1,000×, served as the template for the second PCR, which produced overlapping ∼1-kb fragments (primers available from the authors on request). In addition, we selected other, previously published NRY PCR primers to test for binary markers (Underhill et al. 2001). The PCR products were sequenced in an ABI3700, sequences were analyzed with Consed program (Gordon et al. 1998) for SNPs, and SNPs thus found were validated by visual inspection.
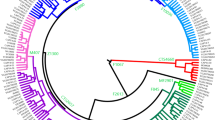
The phylogenetic relationships of the haplogroups are known (Y Chromosome Consortium 2002). In this paper, the 42 markers (including 13 new binary markers and an additional 29 previously published) define 34 haplogroups (Fig. 1a). Figure1b represents the frequency distributions of the occurrence of the haplogroups within each of Chinese ethnical subgroups in the extensive data set (Su et al. 2000; Underhill et al. 2000; Karafet et al. 2001; Wells et al. 2001; Zerjal et al. 2002; and our work). Figure 1 also includes the formal nomenclature of these haplogroups according to the Y chromosome Consortium (YCC) (Y Chromosome Consortium 2002).

Maximum parsimony phylogeny of Chinese nonrecombinant Y-chromosome (NRY) chromosome binary variation. a Tree was constructed from known phylogenetic relationships (Y Chromosome Consortium 2002), and newly discovered binary markers (numbered with “N” capitaled digitals). New haplogroups are labeled in block numbers. The lower table shows the distribution of haplogroups by Chinese ethnical subgroups. Haplogroup nomenclature used here is indicated, as is the formal YCC nomenclature (Y Chromosome Consortium 2002). b The pie charts represent the frequency distributions of haplogroup occurrence within each Chinese ethnical subgroup. Total numbers of individuals in the respective haplogroups are also indicated following the corresponding haplogroup names
Statistical analysis
For this study we analyzed a total of 4,601 men representing 199 populations/locations from China, Northeast Asia (NE.Asia), South Asia (S.Asia) and Central Asia (C.Asia) including sources surveyed in Su et al. (2000), Underhill et al. (2000), Karafet et al. (2001), Wells et al. (2001), Zerjal et al. (2002), in addition to 76 new samples collected throughout China. We used the nested cladistic analysis (Templeton et al. 1995) to explain any statistically significant association between haplogroups and geography in terms of inferred effects of population history (e.g., contiguous range expansion, long distance colonization and fragmentation) and/or population structure (e.g., recurrent gene flow restricted by IBD and long distance dispersal). Nested cladistic analysis was carried out using GeoDis version 2.0 (Posada et al. 2000).
Results and discussion
New binary markers were found
Although the sequences investigated have been DHPLC-scanned for SNPs (Underhill et al. 2000), we discovered a relatively high rate of previously unknown SNPs (13 new compared to 29 previously known). This finding that the Chinese population has many SNPs of restricted global occurrence may indicate that the Chinese haplogroups are partially independently evolved. We refer to these new SNPs as markers N1–N12 and N14. As N6 and N7 indicate the same haplogroup and N14 indicates the same haplogroup as M120, the 13 new markers define only 11 new haplogroups. Of the new markers, eight are found in the lineages M214/M175, which Y Chromosome Consortium (2002) defined as haplogroup O. In Underhill et al. (2001), the haplogroup that carry M214 also carry M175. Here we report a new haplogroup that has M214 but not M175. Thus, we propose redefining haplogroup O as the M214 lineage according to the nomenclature system proposed by Y chromosome Consortium (YCC) (Y Chromosome Consortium 2002). This new haplogroup that has M214 but not M175, is defined as haplogroup O*, and the M175 lineage is redefined as haplogroup O1 (see Fig. 1a).
Nested cladistic analysis of Chinese Y-chromosome haplogroups
Figure 2 displays the nested cladogram for the 62 haplogroups and part of paragroups (as examples of a total of 51 paragroups) generated according to the nesting rules given in Templeton et al. (1987) and Templeton and Sing (1993). This nesting methodology produced 32 one-step clades, 13 two-step clades, 6 three-step clades, 3 four-step clades, and a single five-step clade that encompassed the entire cladogram. Here the higher-level clades that included only one subclade (i.e., itself) are denoted as lower-level subclades and not counted again (e.g., Clade 2–9). The status (interior/tip) of non-monophyletic haplogroups from published data in the nested cladogram is also hard to determine, and similar problems have mentioned in recent studies (Weale et al. 2003). To solve this problem, we have found the following procedure feasible. In this paper, the criterion by which we analytically discriminate between “interior” and “tip” is inferred from the definition given in Posada et al. (2000). The non-monophyletic haplogroups which have the obviously derived/younger haplogroups (such as haplogroup O1a* which has the obviously derived haplogroup O1a2 in Clade 1–18), are here designated as the “interior”. On the other hand, the paragroups defined by the bracketing system (Y Chromosome Consortium 2002), which do not have the explicitly derived haplogroups (such as paragroup N*(xN3) in Clade 1–17), we all regard as “tip”. All “tip” paragroups mentioned above are placed in the corresponding level clades on the basis of their known phylogenetic relationships, and are only analyzed as a part of a source for the higher-level clades, ignoring their relationships with other subclades within the low-level clades. For example, paragroup N*(xN3) is placed as tip in clade 1–17, and is only analyzed as a part of the data of the corresponding higher-level clade 2–9, ignoring the mutual relationships with other subclades (haplogroup N1 and N3) within clade 1–17.

Nested cladistic design for 62 NRY haplogroups and part of polyphyletic paragroups. Hollow circles represent haplogroups that were missing in this sample of Y-chromosomes. New haplogroups are remarked with diamond. The other haplogroups are in accordance with the formal YCC nomenclature, with the exception of haplogroups in Clade 4–2 (haplogroup O) for redefining the M214 lineage in this paper (for example, haplogroup O1a2 in the present designation corresponding to haplogroup O1b in the formal YCC nomenclature). Polyphyletic paragroups remarked with star are the examples and the similar ones discussed in “Results and discussion”
The nested cladistic analysis indicated statistically highly significant associations between clades and geographic locations for 26 of 55 nesting clades. The null hypothesis of no association between the haplogroup/paragroup and geography rejected, our analysis continued by generating specific explanatory inferences involving population history and/or structure considerations. With the aid of a key published on the GeoDis 2.0 Web site (Posada et al. 2000), we were able to infer the probable causes for these 26 patterns (Table 2). Population structure inferences indicated not only restricted gene flow with isolation by distance (IBD; n=4 inferences), but also our sampling design inadequate to discriminate between isolation by distance (short distance movements) and long distance dispersal processes (IBD/LDD; n=14). Population history events included contiguous range expansion (CRE; n=3), past fragmentation (PFR; n=2) and sampling design inadequate to discriminate between contiguous range expansion and long distance colonization (CRE/LDC; n=3). It is clear that both population structure processes and history events indicated vast migrations and extensive ethnical admixture, and have played important roles in shaping Chinese population.
In this survey we found that the main haplogroups D, C, O, L, P were represented in our Chinese sample (Fig. 1a). This agrees with, but also expands on, previous studies that only reported haplogroup O in smaller Chinese samples (Underhill et al. 2000; Karafet et al. 2001; Wells et al. 2001).
Haplogroup D has been reported primarily in Japan and Tibet (Su et al. 2000; Underhill et al. 2001) and also been found in subgroup West (Karafet et al. 2001; Wells et al. 2001). We report it in two additional Chinese ethnic-minority subgroups (subgroup North and South) as well as subgroup Tibet (Fig. 1a). The NCA analysis for haplogroup D revealed one episode of restricted gene flow with isolation by distance (IBD) associated with Clade 2–3 (subgroup West, South, Han, Northeast Asia and South Asia to North, Tibet) and evidence for a contiguous range expansion associated with Clade 1–5 (subgroup Tibet to North).
Haplogroup C was found in Central Asia, South Asia, and East Asia (Karafet et al. 2001; Underhill et al. 2001; Wells et al. 2001; Zerjal et al. 2002). Here it was reported in all Chinese ethnic-minority subgroups and accounted for a high proportion of subgroup North especially. The NCA analysis for haplogroup C indicated one episode of IBD associated with Clade 2–2 (subgroup North, West, South, Northeast Asia, and South Asia to Tibet, Han and Central Asia).
Haplogroup O is dominant (∼65%) in our Chinese sample, as found in previous studies. It is dominant in the Chinese majority Han as well as an important component in all Chinese ethnic-minority subgroups. One episode of IBD was revealed by the NCA analysis for haplogroup O associated with Clade 2–13 (South Asia, Northeast Asia and all Chinese subgroups to Central Asia). The NCA analysis also gives evidence for other five population structure processes (i.e., sampling design inadequate to discriminate between isolation by distance versus long distance dispersal processes) (IBD/LDD) at different levels in the cladogram: South Asia to subgroup Han, associated with Clade 1–18; subgroup Tibet to South, Han and South Asia, associated with Clade 1–19; subgroup West to other four Chinese subgroups and all around China, associated with Clade 2–10; subgroup North, West and all around China to Tibet, South and Han, associated with Clade 3–4; and among all Chinese subgroups and all around China, associated with Clade 3–3.
There was also evidence for population history events for haplogroup O revealed by the above NCA analysis: a contiguous range expansion (CRE) detected in Clade 1–22 (Northeast Asia, Han to four Chinese ethnic-minority subgroups, and South Asia); two past fragmentations (PFR) associated with Clade 2–11 (Tibet to other four Chinese subgroups and all around China) and Clade 2–12 (subgroup Han to Tibet and South); and also another population history event (the sampling design inadequate to discriminate between contiguous range expansion and long distance colonization) (CRE/LDC) detected in Clade 1–26 (West to other three Chinese ethnic-minority subgroups, Central Asia and Northeast Asia).
Evolution and migration history of Chinese population
Despite China’s insular historical image, contacts with other parts of Asia have always been extensive and varied. Here we evaluate the implications of our new genetic data, when compared with existing geographical and historical insight, for understanding the evolution and migration history of the ancestors of today’s Chinese. Based on Chinese ethnological studies (Rene 1998; Fei 1999; Wei and Wang 2000) combined with the NRY variations by geographical locations and ethnical groups found, as mentioned above, we classified the Chinese ethnical minorities into four subgroups (Table 1).
Our first subgroup includes ten populations who live in the agriculturally productive lowlands and river valleys in South China, so we place them together with subgroup South. This subgroup’s NRY lineages are overwhelmingly derived from M175 (i.e., haplogroup O1). The haplogroup O* with M214 but not M175 was not found in Han Chinese and subgroup South but among other three Chinese ethnic-minority subgroups in five samples, constituting 7% of the total and 10% of the M214-containing (haplogroup O) samples in our study. Since haplogroup O*, apparently the immediate ancestor lineage, has not been reported outside China and all of the M175 lineages derived from haplogroup O* exist in China, we tentatively conclude that M214 originated in China and that haplogroup O mainly evolved in China. This also gives another clue to the evolution of the Han Chinese and the subgroup South. The sublineage containing M175 spread from West/North with the migration of Chinese ancestors, became widespread throughout Han and the related subgroup South, and eventually completely replaced other M214 sublineages (Fig. 3). The NCA analysis for haplogroup O also provides some clues to this presumption including a population history event (CRE/LDC) associated with Clade 1–26.

Samples and haplogroups distribution and hypothesized migration routes in China. Chinese ethnical subgroups: Diamond, subgroup Tibet; Circle, subgroup North; Upward triangle, subgroup West; Downward triangle, subgroup West; and Star, Majority Han. Haplogroups: Black, haplogroup D; Blue, haplogroup C; Yellow, haplogroup O1; Red, haplogroup O*; and Green, haplogroup L, P. Arrows: hypothesized original migration routes of Chinese population mentioned in “Results and discussion”
People in haplogroup O are found all over China and surrounding countries. Sixty-five percent of our samples are in haplogroup O, while in the Chinese majority Han the proportion is even higher (Underhill et al. 2000). Based on the fact revealed in their data that southern populations are much more polymorphic than northern populations (Tibetan also included), which have only a subset of the southern haplogroups, Su et al. (1999) have previously proposed the hypothesis that the Chinese originated in south China. But with the continuous accumulation of related data, this hypothesis is facing challenge (Karafet et al. 2001). Our study indicates that haplogroup O is by and large similarly distributed throughout China. This even applies to haplogroup O1b1, corresponding to haplogroup H11∼H12 in Su et al. (1999) (i.e., haplogroup O2a; Y Chromosome Consortium 2002), to one sample in subgroup North (Karafet et al. 2001), and to 14 samples in subgroup Tibet (Su et al. 2000; Karafet et al. 2001 and this paper), further questioning the southern origin hypothesis. On the other hand, as the haplogroup O1a2 (i.e., haplogroup H10; Su et al. 1999), is so far found only in five samples from Taiwan (Underhill et al. 2000), and only as a sublineage of the M175 lineage, the support for the south-origin hypothesis appears less convincing than our West/North-origin scenario inferred from the distribution of the apparent immediate ancestor lineage haplogroup O*.
The second subgroup includes ten populations in and around the Tibetan plateau, which we call together with subgroup Tibet. The particular high-altitude pastoral lifestyle of the Tibetan plateau would have limited interbreeding with surrounding people and encouraged genetic distinctiveness. It has a high incidence of haplogroup D and O in our data, supporting the ethnological theory (Ge et al. 1997; Chu et al. 1998; Su et al. 2000) that the Tibetan ancestors originated from the Di-Qiang population in the upper Yellow River region northeast of Tibet, as is also confirmed in Chinese historiography. Thus as inferred from our analysis for the distribution of haplogroup O*, the ancestors of the Sino-Tibetan population were originally from West/North China. It was also noticed that three samples of haplogroup O* surveyed belong to this subgroup, which may be an indication of a remnant of ancestors from West/North China in the contemporary gene pool of subgroup Tibet, as further suggests that the Di-Qiang contained both M214 and M174 elements before their migration into Tibetan plateau began. About 5,000–6,000 years ago, one brave Di-Qiang branch left their Yellow River homeland, marching westwards and then southward, leading to central Tibet forming the Zang and Kham (both Tibetan), and another branch migrated southward to the east of Tibet, forming the Qiang population in northern Sichuan. Additional migration to the southwest of Sichuan left Yi (Lolo) and Primi, further Di-Qiang formations spread as far as Yunnan and Guizhou provinces where they have a substantial admixture with other Chinese populations (Fig. 3).
Subgroup North constitutes the third subgroup, and includes traditionally nomadic populations in the Mongolian highland as well as hunting and gathering populations. It includes four main ethnical subfamilies: Mongol (including Mongolian and Daur) belonging to the Altaic language family (Mongolian) and Manchu (including Manchu, Hezhe and Xibo) also belonging to Altaic (Manchu-Tungus), which probably both originated in the Ergun and Nengjiang river basins in northeastern China; Yugur, part of which probably the descendants of Mongols; and Korea which language family is undetermined though somewhat similar to Altaic. Genetically, it has a high incidence of haplogroups C and O. In historical times, this family has been associated with nomad incursions and contrasted with the lowland agricultural society centered in the Central Plain. This subgroup’s predominantly non-agrarian lifestyle helped to maintain its distinct genetic identity in spite of interbreeding with other Chinese subgroups.
The M130/M216 lineage (i.e., haplogroup C) is typical of the inhabitants of the Eurasian temperate steppe, and has been found in 55% of Mongolians (Wells et al. 2001). In our data, a slight concentration of haplogroup C is found in 44% of our subgroup North subjects. Conditions in most of the Eurasian temperate steppe were harsh and population densities low. Research shows that most of the historical nomadic tribes originated in the edge of the prairie, especially in the Heilong (i.e., Amur) river region and Altai region, where hunting and gathering could support primeval life before pasture techniques were developed (Rene 1998). Westward and southward dispersal occurred in times of population pressure after nomadic groups were forced out (e.g., the Hun, Turki, and Uyghur) or conquered agricultural states (e.g., the Mongols), which both impelled more vast migrations and more extensive ethnical admixture that swept the whole Eurasia.
The fourth subgroup, subgroup West, includes six Muslim minorities mainly from West China, which have haplogroups from central Asia and the Near East, and is mostly distributed along the Silk Road. As the route of the Silk Road, West China has long been a meeting place of East and West Asian people, so a distinctive genetic heritage is to be expected. Haplogroup L, previously found in South Asia and Central Asia (Karafet et al. 2001; Underhill et al. 2001; Wells et al. 2001; Zerjal et al. 2002), exclusively exits in subgroup West and is absent among other three Chinese ethnic-minority subgroups as well as Han. M45, a mutation that is globally widespread but generally absent in China, corresponding to haplogroup P, is also found here. Here we also classified Hui in this subgroup, although which are distributed throughout China. The Hui trace their descent to traders and artisans from Central and West Asia that migrated to China with the Mongol armies in the 13th century. This subgroup has also a high incidence of haplogroup O (62%) and smaller fractions in haplogroup C. In contrast to other Chinese subgroups, the ethnical admixture between subgroups North and West is more extensive and contacts between them and Central Asia as well as Northeast Asia have been more frequent due to geographical factors and their similar lifestyles. Considering the vast migration in historical times, however, based on existing information it is hard to further infer their earlier historical origin.
The multilayered, multidirectional and continuous Chinese ethnical admixture is exhibited in detail by our nested cladistic analysis. Work on Chinese mitochondrial DNA, Y-chromosome polymorphisms, and STR loci had ever led to conflicting conclusions regarding the origin of Chinese (Chu et al. 1998; Su et al. 1999; Yao et al. 2002). Our results give some new clues to the evolution of Chinese population and their subsequence moving about in this land, which are in accordance with the historical records after agricultural expansion. Wider sampling, additional typing of Y-chromosome, mitochondrial and autosomal markers, and further study of the historical records are likely to throw more light on the study of Chinese ethnical admixture.
Electronic-source information
-
Consed program: http://www.phrap.org/index.html
-
Description of 218 NRY markers: http://www.gene.ucl.ac.uk/anhumgen/Underhill_Appendex_I_2.doc (for published SNPs and primers used in this document)
-
SNPs discovered in this study: http://www.ncbi.nlm.nih.gov/SNP/snp_search.cgi?searchType=byBatch&batch_id=5340, [accession number ss4472667–ss4472678]
-
The GeoDis 2.0 website: http://inbio.byu.edu/Faculty/kac/crandall_lab/geodis.htm
References
Cavalli-Sforza LL (1998) The Chinese human genome diversity project. Proc Natl Acad Sci USA 95:11501–11503
Chu JY, Huang W, Kuang SQ, Wang JM, Xu JJ, Chu ZT, Yang ZQ, Lin KQ, Li P, Wu M, Geng ZC, Tan CC, Du RF, Jin L (1998) Genetic relationship of populations in China. Proc Natl Acad Sci USA 95:11763–11768
Davis RS, Ranov VA (1999) Recent work on the paleolithic of Central Asia. Evol Anthropol 8:186–193
Ding YC, Wooding S, Harpending HC, Chi HC, Li HP, Fu YX, Pang JF, Yao YG, Yu JGX, Moyzis R, Zhang YP (2000) Population structure and history in East Asia. Proc Natl Acad Sci USA 97:14003–14006
Fei X (1999) The pattern of diversity in unity of the Chinese nation. Central University for Nationalities, Beijing
Ge J, Wu S, Chao S (1997) The migration history of China (in Chinese). Fujian People, Fuzhou, China
Gladney DC (1996) Muslim Chinese: ethnic nationalism in the people’s republic. Harvard University, Cambridge, MA
Goebel T (1999) Pleistocene human colonization of Siberia and peopling of the Americas: an ecological approach. Evol Anthropol 8:208–227
Gordon D, Abajian C, Green P (1998) Consed: a graphical tool for sequence finishing. Genome Res 8:195–202
Hammer MF, Karafet TM, Rasanayagam A, Wood ET, Altheide TK, Jenkins T, Griffiths RC, Templeton AR, Zegura SL (1998) Out of Africa and back again: nested cladistic analysis of human Y-chromosome variation. Mol Biol Evol 15:427–441
Hammer MF, Karafet TM, Redd AJ, Jarjanazi H, Santachiara- Benerecetti S, Soodyall H, Zegura SL (2001) Hierarchical patterns of global human Y-chromosome diversity. Mol Biol Evol 18:1189–1203
Karafet TM, Zegura SL, Posukh O, Osipova L, Bergen A, Long J, Goldman D, Klitz W, Harihara S, de Knijff P, Wiebe V, Griffiths RC, Templeton AR, Hammer MF (1999) Ancestral Asian source(s) of new world Y-chromosome founder haplotypes. Am J Hum Genet 64:817–831
Karafet TM, Xu L, Du R, Wang W, Feng S, Wells RS, Redd AJ, Zegura SL, Hammer MF (2001) Paternal population history of East Asia: sources, patterns, and microevolutionary processes. Am J Hum Genet 69:615–628
Lell JT, Sukernik RI, Starikovskaya YB, Su B, Jin L, Schurr TG, Underhill PA, Wallace DC (2002) The dual origin and Siberian affinities of native American Y chromosomes. Am J Hum Genet 70:192–206
Li S (1987) History of Chinese ethnic groups (in Chinese). Orient publishing Center, Beijing
Posada D, Crandall KA, Templeton AR (2000) GeoDis: a program for the cladistic nested analysis of the geographical distribution of genetic haplotypes. Mol Ecol 9:487–488
Qamar R, Ayub Q, Mohyuddin A, Helgason A, Mazhar K, Mansoor A, Zerjal T, Tyler-Smith C, Mehdi SQ (2002) Y-chromosomal DNA variation in Pakistan. Am J Hum Genet 70:1107–1124
Rene G (1998) The empire of the steppes [Empire des steppes, in Chinese]. Commercial, Beijing
Su B, Xiao J, Underhill PA, Deka R, Zhang W, Akey J, Huang W, Shen D, Lu D, Luo J, Chu J, Tan J, Shen P, Davis R, Cavalli-Sforza LL, Chakraborty R, Xiong M, Du R, Oefner P, Chen Z, Jin L (1999) Y-Chromosome evidence for a northward migration of modern humans into Eastern Asia during the last ice age. Am J Hum Genet 65:1718–1724
Su B, Xiao C, Deka R, Seielstad MT, Kangwanpong D, Xiao J, Lu D, Underhill PA, Cavalli-Sforza LL, Chakraborty R, Jin L (2000) Y chromosome haplotypes reveal prehistorical migrations to the Himalayas. Hum Genet 107:582–590
Tajima A, Pan I-H, Fucharoen G, Fucharoen S, Matsuo M, Tokunaga K, Juji T, Hayami M, Omoto K, Horai S (2002) Three major lineages of Asian Y chromosomes: implications for the peopling of east and southeast Asia. Hum Genet 110:80–88
Templeton AR, Sing CF (1993) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. IV. Nested analyses with cladogram uncertainty and recombination. Genetics 134:659–669
Templeton AR, Boerwinkle E, Sing CF (1987) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. I. Basic theory and an analysis of alcohol dehydrogenase activity in Drosophila. Genetics 117:343–351
Templeton AR, Routman E, Phillips CA (1995) Separating population structure from population history: a cladistic analysis of the geographical distribution of mitochondrial DNA haplotypes in the tiger salamander, Ambystoma tigrinum. Genetics 140:767–782
Thomson R, Pritchard JK, Shen P, Oefner PJ, Feldman MW (2000) Recent common ancestry of human Y chromosomes: evidence from DNA sequence data. Proc Natl Acad Sci USA 97:7360–7365
Underhill PA, Jin L, Zemans R, Oefner PJ, Cavalli-Sforza LL (1996) A pre-Columbian Y chromosome-specific transition and its implications for human evolutionary history. Proc Natl Acad Sci USA 93:196–200
Underhill PA, Shen P, Lin AA, Jin L, Passarino G, Yang WH, Kauffman E, Bonne-Tamir B, Bertranpetit J, Francalacci P, Ibrahim M, Jenkins T, Kidd JR, Mehdi SQ, Seielstad MT, Wells RS, Piazza A, Davis RW, Feldman MW, Cavalli-Sforza LL, Oefner PJ (2000) Y chromosome sequence variation and the history of human populations. Nat Genet 26:358–361
Underhill PA, Passarino G, Lin AA, Shen P, Mirazon Lahr M, Foley RA, Oefner PJ, Cavalli-Sforza LL (2001) The phylogeography of Y chromosome binary haplotypes and the origins of modern human populations. Ann Hum Genet 65:43–62
Weale ME, Shah T, Jones AL, Greenhalgh J, Wilson JF, Nymadawa P, Zeitlin D, Connell BA, Bradman N, Thomas MG (2003) Rare deep-rooting Y chromosome lineages in humans: lessons for phylogeography. Genetics 165:229–234
Wei DC, Wang RL (2000) Migration and evolution history of Chinese ethnic groups (in Chinese). Hubei People’s publishing house, Wuhan
Wells RS, Yuldasheva N, Ruzibakiev R, Underhill PA, Evseeva I, Blue-Smith J, Jin L et al (2001) The Eurasian heartland: a continental perspective on Y-chromosome diversity. Proc Natl Acad Sci USA 98:10244–10249
Wong HM, Dajani AA (1988) Islamic frontiers in China. Scorpion, London
Yao YG, Kong QP, Bandelt HJ, Kivisild T, Zhang YP (2002) Phylogeographic differentiation of mitochondrial DNA in Han Chinese. Am J Hum Genet 70:635–651
Y Chromosome Consortium (2002) A nomenclature system of the tree of human Y-chromosomal binary haplogroups. Genome Res 12:339–348
Zerjal T, Wells RS, Yuldasheva N, Ruzibakiev R, Tyler-Smith C (2002) A genetic landscape reshaped by recent events: Y-chromosomal insights into central Asia. Am J Hum Genet 71:466–482
Acknowledgements
We are grateful to the DNA donors and the investigators who helped in collecting the samples. We thank Miss Liang Li (undergraduate student, Beijing University, Beijing) for experiment assistant, and Dr Geir Skogerbo (Visiting Scientist, Chinese Academy of Sciences, Beijing) for providing helpful discussion and critical review of our manuscript. This work was supported by Chinese Academy of Sciences Grant KSCX2-2-07, the National High Technology Development Program (973) of China under Grant No. 2002CB713805, and Beijing Science and Technology Commission Grant H010210010113.
Author information
Authors and Affiliations
Corresponding author
Additional information
The first two authors contributed equally to this study.
Rights and permissions
About this article
Cite this article
Deng, W., Shi, B., He, X. et al. Evolution and migration history of the Chinese population inferred from Chinese Y-chromosome evidence. J Hum Genet 49, 339–348 (2004). https://doi.org/10.1007/s10038-004-0154-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-004-0154-3
Keywords
This article is cited by
-
Genetic analysis of 42 Y-STR loci in Han and Manchu populations from the three northeastern provinces in China
BMC Genomics (2023)
-
Population genetic study of 17 Y-STR Loci of the Sorani Kurds in the Province of Sulaymaniyah, Iraq
BMC Genomics (2022)
-
Genetic Architecture of Southeast-coastal Indian tribal populations: A Y-chromosomal phylogenetic analysis
Egyptian Journal of Forensic Sciences (2019)
-
Prediction of Y haplogroup by polymerase chain reaction-reverse blot hybridization assay
Genes & Genomics (2019)
-
The rs2147578 C > G polymorphism in the Inc-LAMC2–1:1 gene is associated with increased neuroblastoma risk in the Henan children
BMC Cancer (2018)
