
Abstract
We report three possibly disease-causing point mutations in one of the inner-ear-specific genes, KIAA1199. We identified an R187C mutation in one family, an R187H mutation in two unrelated families, and an H783Y mutation in one sporadic case of nonsyndromic hearing loss. In situ hybridization indicated that the murine homolog of KIAA1199 mRNA is expressed specifically in Deiters’ cells in the organ of Corti at postnatal day zero (Pn) P0 before the onset of hearing, but expression in those cells disappears by day P7. The signal of KIAA1199 was also observed in fibrocytes of the spiral ligament and the spiral limbus through to P21, when the murine cochlea matures. Thus, the gene product may be involved in uptake of potassium ions or trophic factors with a particular role in auditory development. Although the R187C and R187H mutations did not appear to affect subcellular localization of the gene product in vitro, the H783Y mutation did present an unusual cytoplasmic distribution pattern that could underlie the molecular mechanism of hearing impairment. Our data bring attention to a novel candidate for hearing loss and indicate that screening of mutations in inner-ear-specific genes is likely to be an efficient approach to finding genetic elements responsible for deafness.
Similar content being viewed by others


Introduction
Hereditary hearing loss is a highly heterogeneous sensory disorder in genetic terms. So far, more than 70 deafness loci have been mapped on human chromosomes, and 29 genes controlling hearing have been identified (Hereditary Hearing Loss home page). The cochlea is composed of structurally and functionally diverse cell types; many forms of deafness involve defects in the hair cells that work as mechanosensors either directly or indirectly. The deafness genes already identified encode a wide variety of proteins, including gap junctions, ion channels, extracellular matrix proteins, transcription factors, and cell/cell adhesion molecules. Obviously, identification of genes that confer proper function of a cochlear-cell type or site is central to an understanding of the molecular mechanism of normal development and maintenance of hearing.
To supplement the candidate-gene approach based on linkage analyses, alternative strategies for reaching that goal rely on recent developments, such as inner-ear cDNA libraries, RT-PCR tests for candidate genes, and expression profiling of inner-ear genes by microarray technologies to identify genes specifically or preferentially expressed in the ear. These advances can also reduce the number of candidates to be tested within a mapped deafness locus. Using a cDNA microarray for analysis, we recently reported CRYM (MIM 123740) as one candidate for nonsyndromic deafness from a set of 52 genes specifically or exclusively expressed in the human inner ear (Abe et al. 2003). More information about genes that are specific to or preferentially expressed in the inner ear will revolutionize our knowledge of the molecular processes involved in auditory function.
In this study, we screened these 52 genes one by one for mutations in patients with hearing loss and identified the disease-associated mutations of KIAA1199 protein gene (KIAA1199), as it shows no significant homology to any known function domain. We also show the unique localization and development of KIAA1199 in the mouse cochlea.
Materials and methods
Candidate gene selection and cloning of the full-length KIAA1199
Among a set of 52 genes specifically or preferentially expressed in the in cochlea and vestibule through cDNA microarray analysis (Abe et al. 2003), we screened each gene using direct sequencing of the coding exons PCR products. We focused on the KIAA1199 protein gene (KIAA1199), one of a collection of genes that showed a high level of expression in the inner ear, and it was selected for our further analysis. We established the full-length KIAA1199 cDNA sequence derived from the original entry XM_051860 by using RT-PCR experiment and direct sequencing. Entry XM_051860 of KIAA1199 mRNA sequence has been replaced in the GenBank. Sequence of major transcript of KIAA1199 has been submitted to GenBank (accession number AB103330). To confirm our cDNA microarray data, we performed semiquantitative RT-PCR experiments using cDNAs derived from the vestibule and from the 29 normal tissues using single-round amplified RNAs, as reported previously (Abe et al. 2003). Primers used to amplify specific KIAA119 products were F: CTAATGCAAGGGTCTCACAC and R: ACCAGCTATTTCGGCAACAG; the PCR proceeded for 30 cycles of 95°C for denaturing, 55°C for annealing, and 72°C for extension. The PCR products of 348 bp lengths were electrophoresed on a 2% agarose gel and visualized by UV light.
Northern-blot analysis
A gel-purified 505 nt cDNA fragment of KIAA1199 mRNA (GenBank accession number AB103330, nucleotides 6495–6999), the same product as that spotted onto the cDNA microarray slides, was used to probe a northern blot. A human multitissue blot (Clontech, Palo Alto, CA, USA) hybridized with the DNA probe that labeled with random-primed [32P]dCTP using Rediprime DNA labeling system (Amersham Life Science, Cleveland, OH, USA). The condition of hybridization and washing was carried out as previously described (Koyama et al. 1995).
Patients
We described earlier the ascertainment of 192 Japanese patients with or without families in which probands sustained congenital or childhood-onset bilateral nonsyndromic sensorineural hearing loss (Abe et al. 2003). With the exception of hearing impairment, no patient showed untoward neurological signs. Written informed consent was obtained from all family members who participated in this study. Pure-tone audiometry was systematically performed for each individual; in younger children, auditory function was assessed by the auditory brainstem response (ABR) and conditioned orientation reflex audiometry. The control group consisted of 96 unrelated Japanese volunteers whose normal hearing was confirmed by auditory testing.
Mutation screening of KIAA1199
We screened DNA of one affected person from each family. The exon/intron structure of KIAA1199 was generated using a combination of nuclei acid sequence database homology search from NCBI and BAC sequence (GenBank accession number AC027808). Twenty-seven DNA fragments encompassing the 28 coding exons of KIAA1199 and their flanking sequences were PCR amplified from genomic DNA (10–20 ng) using the 27 primer pairs listed in Table 1. PCR products were sequenced by using 27 additional internal primers (Table 1). Details of the methods for PCR amplification and sequencing analysis were as described elsewhere (Abe et al.2003). PCR products were sequenced using 27 additional internal primers (Table 1).
Cloning of mouse homolog of KIAA1199
To determinate mouse KIAA1199 cDNA sequence, human KIAA1199 cDNA sequence was aligned to publicly available mouse cDNA sequences through the BLAST searches of the NCBI database. Four ESTs, BB62981, BB620830, AY007815, and NM_030728, have shown high identity with more than 85% of nucleotides. The complete mouse KIAA1199 homolog cDNA sequence was determined and confirmed by ESTs connection PCR and direct sequencing, and have been submitted to GenBank (GenBank accession number AB103331).
In situ hybridization
A 388 nt fragment spanning part of the murine counterpart of KIAA1199 (GenBank accession number AB103331, nucleotides 3721–4108), was cloned into pBlueScript SK(-) vector (Stratagene, Cedar Creek, TX, USA). Probes were prepared by in vitro transcription of linealized plasmid using T7 polymerase for the generation of sense and antisense RNA probes. Mice at postnatal days (Pn) P0, P7, and P21 were dissected prior to fixation in10% neutral formalin. Tissues were embedded in paraffin, and then tissue sectioning was carried out through the midmodiolar plane to provide for the well-defined morphological structure of the auditory apparatus. Serial tissue sections (6 µm) were hybridized with digoxigenin-labeled RNA probes as described before.
Generation of a HA-tagged KIAA1199 protein construct and mutagenesis
Wild-type KIAA1199 cDNA fragment by adding a 3’-tag encoding the HA epitope, excluding the predicted signal peptide (codon, 1–30), was generated by PCR using the following primers: sense 5’-CGTGGTACCCAGACCGTGCATCATGGGAGCTGCTGGGAGGCAGGACTTC -3’, and antisense 5’-AAGCTCGAGTCAAGCGTAGTCTGGGACGTCGTATGGGTACAACTTCTTCTTCTTCACCACAGG –3’, containing KpnI and XhoI sites. This amplicon was purified and digested with KpnI and XhoI, and subcloned into pCDNA 3.1(+) mammalian expression vector (Invitrogen, San Diego, CA, USA). Three R187C, R187H, and H783Y mutants were created by an inverse PCR reaction using 5’phosphorelated primers with their 3’ends facing away each other, containing the desired mutations. PCR was performed using KOD(+) DNA polymerase (TOYOBO, Tokyo, Japan) on wild-type KIAA1199 plasmid template. To circularize, the DNA fragments were ligated. R187C primers: F 5’- GTGGAGTTATTGTTCATGTCATCGACC-3’, R 5’- AGTGGCCCCAGCTCCTTTCA-3’; R187H primers: F 5’-TGGAGTTATTGTTCATGTCATCGAC-3’, R 5’-TGGTGGCCCCAGCTCCTTTCAA-3’; H783Y primers: F 5’-TACTTCATTGCCTACAAGAACCAGGACCA-3’, R 5’-TCTGATGATGGCCGGCTCCCGGGGCT-3’. All constructs were then verified by direct sequencing.
Immunofluorescence
Transfections were performed on COS-7 cells using FuGENE6 (Roche Molecular Biochemicals, Indianapolis, IN, USA) diluted in OPTIMEM medium (Gibco BRL, Grand Island, NY, USA) according to the manufacturer instructions. 5×104 cells cultured on in 2-well glass chamber slides were transfected with 1 µg of each construct for immunofluorescence experiments. At 24 h, 48 h, 72 h, and 96 h after transfection, cells were fixed and treated as described elsewhere (Tsujikawa et al. 1999). They were first incubated overnight at 4°C with mouse anti-HA tag antibody (1:1000) (Santa Cruz Biotechonology, USA), followed by the FITC-conjugated goat anti-mouse IgG (1:3000) (ICN/Cappel, Costa Mesa, CA, USA) for 60 min at 25°C. Slides were examined by means of fluorescence microscopy.
Results
Expression of KIAA1199
KIAA1199 (GenBank accession numbers XM_051860 and AB103330) showed a high level of expression in the inner ear (Cy3/Cy5 ratio: 56.54 in the cochlea, 22.55 in the vestibule) by cDNA microarray analysis (Abe et al. 2003). We confirmed predominant expression of KIAA1199 in the inner ear by means of semiquantitative RT-PCR experiments (data not shown). Northern-blot analysis of multiple human tissues using a partial KIAA1199 cDNA probe yielded a transcript approximately 7 kb long that was expressed only in prostate and testis (data not shown).
Genomic and predicted protein structure of KIAA1199
Comparison of cDNA sequences (GenBank accession number AB103330) with archived genomic sequences (GenBank accession number AC027808) revealed that the gene spans approximately 170 kb and consists of 29 exons (exons 2–29 corresponding to the coding elements). The cDNA sequence contains a predicted 4083-bp ORF encoding a 1361-aa protein, and the PSORT II program (http://psort.ims.u-tokyo.ac.jp/form2.html) predicted that its N-terminal 30 amino acid portion was a cleavable signal peptide. The molecular weight of the mature 1331 amino acid protein was calculated to be 150 kDa. A BLAST search (http://www.ncbi.nlm.nih.gov/BLAST/) revealed 38% identity (63% similarity) to transmembrane protein 2 (TMEM2; GenBank accession number NP_037522, MIM 605835), which has a wide tissue distribution that includes human fetal cochlea (Scott et al. 2000). Part of the gene product (aa 55–155) revealed 38% identity (57% similarity) to PKHD1 protein (fibrocystin; GenBank accession number NP_733842), whose mutant forms are responsible for polycystic kidney and hepatic disease 1 (MIM 606702). Analysis with the ProDom program (http://prodes.toulouse.inra.fr/prodom/2002.1/html/home.php) identified one region of homology (aa 187–278) with O-linked mannose beta1,2-N-acetylglucosaminyltransferase (UDP-GLCNAC; GenBank accession number NP_001373), and with FAM3-family proteins such as human FAM3A (GenBank accession number P98173) and 3B (GenBank accession number P58499), mouse FAM3B (GenBank accession number Q9D309), human FAM3C (GenBank accession number Q92520) and 3D (GenBank accession number AAH15359), and hypothetical protein M70.4 of Caenorhabditis elegans (GenBank accession number AAK71390). No apparent functional domain or motif was predicted by any of the multiple computer programs we invoked.
KIAA1199 mutations analyzed in this study
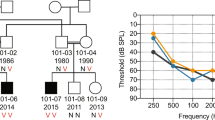
We identified nine DNA variants, including six that would alter the predicted amino acid sequence of KIAA1199, as summarized in Table 2: six missense alterations (R187C, R187H, H783R, H783Y, V1109I, and P1169A) and three synonymous substitutions (L532L, P619P, and D800D). We observed no R187C, R187H, H783Y, or V1109I in any of the 192 chromosomes from the control group without hearing loss, but R187H was found in two unrelated families with hearing-impaired members, suggesting that four missense (R187C, R187H, H783R, or V1109I) mutations may be potentially disease causing. We also detected eight SNPs in intronic regions (Table 3). To clarify whether pathogenic changes might result from rare genetic variations, we investigated segregation in some members of family 69. In family 69 (Fig. 1A), an R187C allele was derived from the unaffected mother and an L532L synonymous substitution from the unaffected father. The DNA sample from other family members was not available.

Detection of KIAA1199 mutations. A Three missense mutations in four probands (arrows), two with family history (families 69 and 189) and two sporadic cases with nonsyndromic hearing loss (families 14 and 70). Sequence chromatograms from wild-type protein are shown. The proband in family 69 and his mother carried a heterozygous 559 C->T transition in exon 5, which results in an arginine to cysteine change (R187C). Another variation, 1594C->T (L532L), was derived from his father. Probands in families 14 and 189 are heterozygous for 560 G->A in exon 5, which replaces arginine with histidine (R187H). The patient in family 70 is heterozygous for a 2347 C->T change in exon 18, which substitutes tyrosine for histidine (H783Y). Since DNA samples were not available from any other family members, we were unable to examine the segregation of mutations any further. B Alignment of KIAA1199 protein, TMEM2, FAM3-family proteins, UDP-GLCNAC, and hypothetical protein M70.4 of Caenorhabditis elegans by means of the ProDom program. Residues between codons 187 and 208 are indicated. An arrow indicates the position of the R187 codon
Distribution of KIAA1199 mRNA in the developing murine cochlea
At P0, the immature period of the organ of Corti in the mouse, KIAA1199 mRNA was localized to the fibrocytes of the spiral ligaments and the spiral limbus in the expansive connective tissue region surrounding perilymph and endolymph (Fig. 2a). In addition, KIAA1199 mRNA was expressed at the Deiters’ cells, which are supporting structures of outer hair cells (OHCs) at their basal pole, as shown in Fig. 2b. KIAA1199 signal in the Deiters’ cells observed at P0 was not present by P7, before the onset of hearing (data not shown). The widespread signal of KIAA1199 mRNA at the connective tissue elements has been restricted, but its expression in the lateral region of the spiral ligament and the fibrocytes of the spiral limbus was maintained at P7 and P21 (data not shown).

In situ hybridization of KIAA1199 mRNA in the mouse cochlea at postnatal day (Pn) P0. Probes were prepared from a 388 nucleotide fragment spanning part of the murine counterpart of KIAA1199 and were labeled with digoxigenin. AS antisense probe, S sense probe. Images were obtained at a magnification of 400×. a Localization of KIAA1199 mRNA in the expansive connective tissue region surrounding perilymph and endolymph (arrows). SLg spiral ligaments (arrowhead). b Specific expression of mouse KIAA1199 mRNA in Deiters’ cells (DC, arrow), which are supporting structures for outer hair cells (OHCs, arrowhead) at their basal poles
Subcellular localization study
To evaluate the effect of three nonsynonymous amino acid changes (R187C, R187H, and H783Y) on functional consequences of these three amino acid changes in the different areas of the protein structures, we performed transiently transfection experiments in the COS-7 cells. At all time points examined after transfection (24 h, 48 h, 72 h, and 96 h, respectively), wild-type KIAA1199 protein was localized predominantly in the cytoplasmic region with some scattered spaces (Fig. 3a). The localization of the R187C and R187H mutants were very similar to that of the wild-type protein (data not shown). The remaining H783Y mutant also showed the cytoplasmic distributed pattern, but the edge and surface showed an irregular and worm-eaten pattern in most transfected cells (Fig. 3b). In cellular extracts of transfected cells with normal and three mutated proteins, ~150 kDa bands representing full-length KIAA1199 protein were detected by western blots using an anti-HA antibody (data not shown).

Subcellular localization of KIAA1199 protein in COS-7 cells transiently transfected with wild-type (a) or H783Y (b) KIAA1199 proteins with HA-tags at their C-termini. Nuclei are counterstained with DAPI (blue). a Localization of wild-type protein (green) in cytoplasm. b Representative pattern of the irregular and worm-eaten appearance of the edge and surface observed in 80% of the cells transfected with the H783Y mutant
Discussion
We screened these 52 genes in this series for mutations in patients with hearing loss. In those experiments, KIAA1199 showed a high level of expression in the inner ear by cDNA microarray analysis (Abe et al. 2003). Although KIAA1199 mRNA is also expressed in the fetal cochlea (Human Cochlear cDNA library and EST database), we did not initially focus on this gene, because its function was unknown and its physical location at chromosome 15q24 was not among the previously known deafness loci. However, since our microarray and RT-PCR data indicated preferential expression of this gene in the inner ear, we selected it for further analysis.
In the present study, we identified nine DNA variants in the KIAA1199 protein gene (Table 2). Segregation study in family number 69 showed that an R187C allele was derived from the unaffected mother and an L532L synonymous substitution from the unaffected father (Fig. 1A). Since DNA samples were not available from any other family members, we were unable to examine the segregation of mutations any further. Hence, to assess the significance of the missense substitutions found in our study population, we examined whether the substitutions had occurred at amino acid residues that were conserved in the murine homologue of KIAA1199 (GenBank accession number AB103331). Among four missense (R187C, R187H, H783Y, or V1109I) mutations, V1109I mutation was not conserved between human and mouse sequence (Table 2); therefore, it seems unlikely to be a disease-causing mutation.
Although the H783R mutation also occurred in the conserved H783 codon, it was observed in ten of the 192 alleles from individuals with normal hearing (Table 2), suggesting that this replacement represent a common polymorphism. On the other hand, the R187 residue is widely conserved among FAM3-family proteins, as well as in UDP-GLCNAC, TMEM2, and hypothetical protein (Caenorhabditis elegans). This conservation over evolutionarily distant species (Fig. 1B) suggests that substitutions at R187 could significantly affect the function of KIAA1199 protein. Phenotypically, the wide variation in the onset of hearing loss and the shape and severity of the audiograms was seen in affected individuals harboring each of three mutations (Table 4). None of them showed abnormal vestibular symptoms.
To investigate the biological significance of this gene product in the auditory system, we performed in situ hybridization experiments using murine mRNA. In the mouse, the counterpart of KIAA1199 is widely expressed in the fibrocytes of the spiral ligament and the spiral limbus at P0 (Fig. 2a). These cell types, rich in ion-transporting molecules, are involved in generating endocochlear potential. At P7, after maturation but before the onset of hearing, and at P21, the expansive signal was confined, but its expression was maintained in the lateral region of the spiral ligament and in fibrocytes of the spiral limbus (data not shown). Maintenance of expression through to the cochlea-matured stage implies that KIAA1199 protein might play a key role in ion homeostasis. We detected a transient signal of KIAA1199 mRNA in Deiters’ cells at day P0 (Fig. 2b), but those signals were no longer observed at P7 (data not shown).
Unlike humans, mice are unable to hear at birth; the first two postnatal weeks are critical for inner-ear development in mice (Rybak et al. 1992). Deiters’ cells, a type of supporting structure in the epithelium of the organ of Corti, are thought to maintain the optimal set point of cochlear amplifiers, such as OHCs or stereocilia to account for the remarkable sensitivity of the cochlea to sound (Flock et al. 1999). Moreover, since Deiters’ cells are closely apposed to the OHCs, extending the phalangeal process apically to contribute to the reticular lamina, these cells may also play an important role as a trophic intermediary for the OHCs (Mothe and Brown 2001). In addition to mechanical and trophic support, these cells contribute to uptake of potassium ions from sensory hair cells and to diffusion of K+ through gap junctions to adjacent supporting cells and fibrocytes (Boettger et al. 2002). Therefore, specific expression in Deiters’ cells within the organ of Corti suggests that KIAA1199 protein may mediate a cochlear epithelial network involving potassium, or expedite passage of other neurotrophic factors between OHCs and the supporting cells before the onset of hearing. Even though no KIAA1199 mRNA is expressed in sensory hair cells, KIAA1199 mutations may induce perturbation of sensory epithelia and lead to auditory deficits; this hypothesis is consistent with the early appearance of Deiters’ cells during development.
We then investigated functional consequences of three nonsynonymous amino acid changes (R187C, R187H, and H783Y) occurring at conserved residues, none of which were detected in any normal controls, by forcing the expression of normal or mutated proteins in COS-7 cells. At all time points examined after transfection (24 h, 48 h, 72 h, and 96 h, respectively), wild-type KIAA1199 protein was localized in the cytoplasmic region with some scattered spaces (Fig. 3a). R187C and R187H mutants indicated patterns very similar to that of the wild-type protein (data not shown), excluding the possibility that the primary detrimental effect was caused in these cases by aberrant subcellular distribution. Therefore, we speculate that both mutations may instead interfere with interaction and cooperation with unknown binding partners. In contrast, the H783Y mutant showed an irregular and worm-eaten pattern in most transfected cells (Fig. 3b), suggesting that incorrect cytoplasmic localization of the H783Y mutant may underlie a molecular process leading to hearing loss.
We have provided here the first report that KIAA1199 protein may be essential for auditory function and that its mutated forms may cause nonsyndromic hearing loss. The mode of inheritance in our test families indicated either autosomal-recessive transmission or simplex cases; we were unable to detect any pathogenic mutations in the other KIAA1199 allele of any affected individual. Possible explanations for penetrance in cases harboring just one missense change include: (1) a second mutation outside the coding sequence, perhaps in an intron or promoter (Lerer et al. 2001), or (2) de novo occurrence in simplex cases. Alternatively, mutations in other genes may be impairing auditory function on their own (Abe et al. 2001; de Brouwer et al. 2003), although the GJB2 gene (MIM 121011) and the mitochondrial gene encoding 12S rRNA (MIM 561000) have already been screened and excluded from this premise (Abe et al. 2003). A full understanding of the consequences of each mutation reported here will require additional study, as will efforts to determine the precise mechanisms operating in the pathophysiology of hearing loss associated with KIAA1199 mutations.
References
Abe S, Kelley PM, Kimberling WJ, Usami SI (2001) Connexin 26 gene (GJB2) mutation modulates the severity of hearing loss associated with the 1555A-->G mitochondrial mutation. Am J Med Genet 103:334–338
Abe S, Katagiri T, Saito-Hisaminato A, Usami S, Inoue Y, Tsunoda T, Nakamura Y (2003) Identification of CRYM as a candidate responsible for nonsyndromic deafness, through cDNA microarray analysis of human cochlear and vestibular tissues. Am J Hum Genet 72:73–82
Boettger T, Hubner CA, Maier H, Rust MB, Beck FX, Jentsch TJ (2002) Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 416:874–878
de Brouwer AP, Pennings RJ, Roeters M, Van Hauwe P, Astuto LM, Hoefsloot LH, Huygen PL, van den Helm B, Deutman AF, M Bork J, Kimberling WJ, Cremers FP, Cremers CW, Kremer H (2003) Mutations in the calcium-binding motifs of CDH23 and the 35delG mutation in GJB2 cause hearing loss in one family. Hum Genet 112:156–163
Flock Å, Flock B, Fridberger A, Scarfone E, Ulfendahl M (1999) Supporting cells contribute to control of hearing sensitivity. J Neurosci 19:4498–4507
Koyama K, Sudo K, Nakamura Y (1995) Isolation of 115 human chromosome 8-specific expressed-sequence tags by exon amplification. Genomics 26:245–253
Lerer I, Sagi, M, Ben-Neriah Z, Wang T, Levi H, Dvorah A (2001) A deletion mutation in GJB6 cooperating with a GJB2 mutation in trans in non-syndromic deafness: a novel founder mutation in Ashkenazi Jew. Hum Mut
Mothe AJ, Brown IR (2001) Expression of mRNA encoding extracellular matrix glycoproteins SPARC and SC1 is temporally and spatially regulated in the developing cochlea of the rat inner ear. Hear Res 155:161–174
Rybak LP, Whitworth C, Scott V (1992) Development of endocochlear potential and compound action potential in the rat. Hear Res 59:189–194
Scott DA, Drury S, Sundstrom RA, Bishop J, Swiderski RE, Carmi R, Ramesh A, Elbedour K, Srikumari Srisailapathy CR, Keats BJ, Sheffield VC, Smith RJ (2000) Refining the DFNB7-DFNB11 deafness locus using intragenic polymorphisms in a novel gene, TMEM2. Gene 246:265–274
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, Nakamura Y (1999) Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nat Genet 21:420–423
Acknowledgements
We thank all of the families that participated in this study. We are also grateful to Yumi Kondoh for help with direct sequencing of DNA. This work was supported in part by Research for the Future Program Grant #00L01402 from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Additional information
Nucleotide sequence data reported herein are available in the DDBJ/EMBL/GenBank databases; for details, see the electronic eatabase section of this article.
Rights and permissions
About this article
Cite this article
Abe, S., Usami, Si. & Nakamura, Y. Mutations in the gene encoding KIAA1199 protein, an inner-ear protein expressed in Deiters’ cells and the fibrocytes, as the cause of nonsyndromic hearing loss. J Hum Genet 48, 564–570 (2003). https://doi.org/10.1007/s10038-003-0079-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-003-0079-2
Keywords
This article is cited by
-
Inhibition of CEMIP potentiates the effect of sorafenib on metastatic hepatocellular carcinoma by reducing the stiffness of lung metastases
Cell Death & Disease (2023)
-
KIAA1199 deficiency enhances skeletal stem cell differentiation to osteoblasts and promotes bone regeneration
Nature Communications (2023)
-
Chitosan oligosaccharide suppresses osteosarcoma malignancy by inhibiting CEMIP via the PI3K/AKT/mTOR pathway
Medical Oncology (2023)
-
CircCEMIP promotes anoikis-resistance by enhancing protective autophagy in prostate cancer cells
Journal of Experimental & Clinical Cancer Research (2022)
-
Loss of REST in breast cancer promotes tumor progression through estrogen sensitization, MMP24 and CEMIP overexpression
BMC Cancer (2022)
