
Abstract
The hamigeran family of natural products has been the target of numerous synthetic efforts because of its biological activity and interesting structural properties. Herein, we disclose our efforts toward the synthesis of hamigerans C and D, unique among the initially isolated members because of their 6-7-5 carbocyclic core. Our approach directly targets this tricyclic motif by sequential Negishi and Heck coupling reactions, yielding an advanced intermediate with all necessary carbons and sufficient functionality poised for completion of the synthesis of these two natural products.
Similar content being viewed by others
Introduction
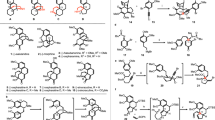
The hamigeran family of natural products was isolated in 1999 from marine sponge Hamigera tarangaensis by Cambie and co-workers1 (Scheme 1). While all four members of the family possess moderate cytotoxicity in P-388 leukemia cells (IC50=31.6, 13.5, 16.0 and 8.0 μM for hamigerans A, B, C and D, respectively), hamigeran B (2) displays 100% in vitro inhibition against both Herpes and Polio viruses.
Owing to this attractive biological activity and the synthetically challenging 6-6-5 tricyclic structure bearing three consecutive stereocenters on the congested A-ring, hamigeran B has been the target of continual efforts of many synthetic organic chemists. Hamigeran B (along with hamigeran A and several analogs) was first synthesized in 2003 by Nicolaou et al.2, 3 and since has succumbed to a number of syntheses,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 including epimeric and formal syntheses. In contrast, at the outset of our studies no synthesis of hamigeran C or hamigeran D (3 and 4), which share a unique 6-7-5 tricyclic core, had been reported. This dearth in syntheses is presumably due to the relative scarcity of methods for the synthesis of seven-membered rings as compared with their six-membered counterparts. Hamigeran D has been synthesized recently by Gao and co-workers15 via a biomimetic skeletal rearrangement from a 6-6-5 tricycle, and thus our strategy of directly targeting the seven-membered ring structure remains unique (vide infra).
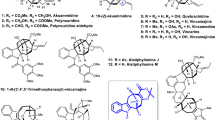
The Stoltz group has developed a method for the asymmetric synthesis of γ-quaternary acylcyclopentenes (Scheme 2, 10) by a retro-aldol/aldol condensation sequence resulting in a two-carbon contraction of vinylogous esters of cycloheptenone (13).16 The versatility of this intermediate and the excellent scalability of this sequence inspired us to pursue the synthesis of natural products with cores containing densely functionalized cyclopentanes with quaternary carbon stereocenters, making hamigerans C and D ideal targets.
Results and discussion
Studies toward the syntheses of hamigerans C and D were initiated during our investigations of the asymmetric synthesis of γ-quaternary acylcyclopentenes.16 From acylcyclopentene 10, we were able to assemble the 6-7-5 carbocyclic core of hamigerans C and D (Scheme 2, 18) in good overall yield over four synthetic steps, featuring two sequential Heck cross-coupling reactions.
Notably lacking on tricycle 18, however, is substitution on the aromatic C-ring and any functional handle on the benzannulated cycloheptyl B-ring of the natural products. The latter is due to the (E)-selectivity in the coupling of 10 with o-iodophenol. The resulting olefin must be hydrogenated to perform the second Heck coupling, resulting in loss of the otherwise useful olefin moiety. As such, we envisioned a retrosynthetic strategy that would allow us to perform the first cross-coupling reaction to yield a (Z)-olefin with an appropriately substituted aromatic ring (Scheme 1, 7). Such a strategy would allow us to obtain a tricyclic intermediate (6) with all of the necessary functional handles in place to advance toward the total syntheses of both of these natural products.
Our first synthetic efforts were directed toward synthesis of Negishi coupling partners 8 and 9. We desired a differentially protected orcinol derivative, as one phenol group would need to be deprotected and converted to an aryl triflate for the planned Heck cross-coupling, while the other phenol would need to be robustly protected as a methyl ether to be revealed late in the synthesis as the phenol present in hamigeran C or the phenol precursor to the oxazine moiety of hamigeran D. Synthesis of the O-methyl-O'-tetrahydro-2-pyranylorcinol 8 was accomplished by dimethylation and monodemethylation of orcinol (19),17 followed by protection as a tetrahydropyranyl ether in 64% yield over three steps (Scheme 3).
The synthesis of (Z)-vinyl iodide 9 was accomplished by a novel sequence from acylcyclopentene 10. Following the established procedure for ketal protection of the enone,16 the resulting diene could be selectively ozonolyzed to yield aldehyde 21 in 71% yield over two steps. However, yields obtained using the (Z)-iodomethylenation procedure reported by Stork and Zhao18 proved unreliable upon several repeated experiments (Table 1).
While mass balance for this transformation was consistent, varying amounts of starting material contaminated by deprotection side products were reisolated, with yields of the desired vinyl iodide 9 ranging from 33 to 67% (Table 1, entry 1). Moreover, the 5:1 ratio of Z:E isomers (as determined by integration of the terminal vinyl proton in the 1H NMR spectrum) could not be separated by chromatography through silica gel, alumina or silver nitrate-impregnated silica gel with a wide variety of eluent combinations. As an unreacted starting material appeared to be a major cause of loss for the yield in this transformation, we considered that, given the quaternary center proximal to the aldehyde of 21, nucleophilic approach by the Wittig reagent was extremely hindered. We reasoned that the Wittig reagent was instead acting as a base, deprotonating the α-position of 21 to yield an unreactive enolate.
Raising the temperature of addition in this reaction to 40 °C and maintaining that temperature throughout the reaction yielded a consistent boost in yield (66–87%) without any detriment to the (Z)-selectivity (Table 1, entry 2). Reaction at 30 °C also appeared to be an improvement, although longer reaction times increased the appearance of side products resulting from deprotection of the ketal (entry 3). By raising the temperature to 50 °C and performing the reaction in benzene, we were able to achieve a good yield, but noted a marked decrease in selectivity from 5:1 to 1.3:1 in favor of the (Z)-olefin (entry 4).
Satisfied with the improved yields obtained by performing the reaction with an addition temperature of 40 °C, we sought to improve the (Z)-selectivity by modifying the nature of the Wittig reagent. We imagined that modification of the aggregation state of the deprotonated Wittig salt by modifying the counterion or base used or by chelation of the counterion by hexamethylphosphoramide could benefit the selectivity of the reaction.
Unfortunately, substitution of sodium hexamethyldisilylamide for lithium or potassium hexamethylphosphoramide (Table 2, entries 2 and 4) both resulted in a decrease in selectivity for the desired (Z)-olefin, as did addition of HMPA (entry 3) or the use of sodium t-butoxide (entry 5). Moreover, tuning of the sterics of the Wittig reagent itself by substituting (iodomethyl)triphenylphosphonium iodide for the less bulky (bromomethyl)triphenylphosphonium bromide (entry 6) or (iodomethyl)tri-n-butylphosphonium iodide (entry 7) also resulted in a decrease in selectivity. To our surprise, the more sterically hindered (iodomethyl)tri-o-tolylphosphonium iodide gave a modest selectivity of 0.9:1 in favor of the undesired (E)-olefin (entry 8). As such, we returned to our initial conditions and proceeded with our synthetic studies.
With the proposed Negishi cross-coupling partners in hand, we began investigations of this reaction. We anticipated being able to affect a selective deprotonation of 8 ortho- to the two ether substituents with n-butyllithium, followed by transmetallation with zinc(II) chloride to afford the Negishi coupling partner for vinyl iodide 9 (Scheme 3).
Gratifyingly, this reaction proceeded in good yield to afford 22 as a single product with respect to the deprotonation of 8, although still as a mixture of olefin isomers. Removal of both acetal and tetrahydropyranyl aceta protecting groups proceeded smoothly using Montmorillonite K-10 clay in methanol at 50 °C, the crude product of which was >95% pure by 1H NMR and could be used directly in the following step. We also found that triflation of the intermediate phenol could be affected using trifluoromethanesulfonyl anhydride and 4-dimethylaminopyridine over a slightly longer reaction time but without loss of yield when compared with the more costly Comins’ reagent (N-(5-chloro-2-pyridyl)bis(trifluoromethanesulfonimide)), furnishing the desired aryl triflate 7 in 55–62% yield over three steps from Negishi coupling partners 8 and 9. At this point, the minor olefin isomer had also been removed over the course of purification and isolation steps, or had otherwise isomerized to the major isomer due to exposure to light and/or heat.
Aryl triflate 7 was subjected to conditions developed for the model Heck reaction (Scheme 2), modified to be run in a microwave reactor, providing tricycle 6 in 88% yield on 0.12 mmol scale (Scheme 3). However, upon scale up, cleavage of the triflate caused a significant decrease in yield, and a large amount of an undesired acylated side product was obtained. Further improvements to this Negishi–Heck sequence, including attempts at performing the steps in a one-pot cascade manner using Pd and Ni catalysis, have been the subject to ongoing research in our group.
With functionalized tricyclic core of hamigerans C and D (6) in hand, we sought to pursue the selective functionalization of the disubstituted olefin. A number of dihydroxylation and epoxidation conditions including AD–mix, stoichiometric osmium tetroxide and m-CPBA all failed to provide the desired product. Epoxidation of the disubstituted alkene 6 was finally affected in a mixture of aqueous bleach and dichloromethane with (R,R)-Mn(salen) epoxidation catalyst 23 to afford epoxide 24 in 89% yield and a 7.5–10:1 ratio of inconsequential diastereomers (Scheme 3).
Treatment of 24 with catalytic gold(III) chloride in dry acetone19 directly yielded protected diol 25 in 74% yield as a single diastereomer, although the stereochemistry of the B-ring substituents has eluded definitive determination. The final carbon of hamigerans C and D could be installed by submitting acetal 25 to Wittig conditions, yielding desired compound 26. Diene 26 represents the most advanced intermediate we have synthesized to date, and completion of the synthesis of hamigerans C and D requires hydrogenation of the diene moiety, deprotection and oxidation of the B-ring diol, bromination and demethylation of the aromatic C-ring. Synthesis of hamigeran C would require installation of the acetoxy group on the B-ring, and completion of hamigeran D would occur by completion of the oxazine D-ring.
In conclusion, we have achieved the synthesis of the 6-7-5 tricyclic core of hamigerans C and D via a unique strategy of directly targeting synthesis of the seven-membered ring via sequential Negishi and Heck cross-coupling reactions. Our targeted synthesis is made catalytically enantioselective by application of our asymmetric alkylation/ring-contraction strategy for synthesis of chiral acylcyclopentenes. Our advanced intermediates possess the requisite carbons and functionality for elaboration to both of these targets, and research into the remaining steps is underway in our group.
For experimental procedures, please see accompanying supporting information.

Structures of the first four identified members of the hamigeran family of natural products, and retrosynthetic analysis of hamigerans C and D.

Previously achieved methods for the synthesis of chiral acylcyclopentenes and elaboration to the carbocyclic core of hamigerans C and D. (a) Pd2(pmdba)3 (1.25 mol %), (S)-t-BuPHOX (12, 3.13 mol%), toluene, 30 °C, 93% yield, 87% ee. (b) LiAlH4, Et2O, 0 °C; 10% aq. HCl, 81%. (c) LiOH, CF3CH2OH, tetrahydrofuran (THF), 60 °C, 91%. (d) Pd(OAc)2 (10 mol%), tetrabutylammonium iodide (TBAI), NEt3, o-iodophenol, dimethylformamide, 100 °C, 90%. (e) Rh(PPh3)3Cl (10 mol%), H2 (1 atm), toluene, 23 °C, 95%. (f) Comins’ reagent, 4-dimethylaminopyridine (DMAP), CH2Cl2, 78%. (g) Herrmann–Beller catalyst (17, 10 mol%), Bu4NOAc, DMA, 115 °C, 77%.

Synthetic efforts toward the total synthesis of hamigerans C and D. (a) Me2SO4, K2CO3, refluxing acetone, 95%. (b) NaH, EtSH, refluxing DMF, 84%. (c) dihydropyran (DHP), p-toluenesulfonic acid, tetrahydrofuran (THF), 81%. (d) neopentyl glycol, pyridinium p-toluenesulfonate (PPTS), toluene, 140 °C, 73%. (e) O3, CH2Cl2, –78 °C; PPh3, –78 °C→23 °C, 97%. (f) ICH2PPh3I, sodium bis(trimethylsilyl)amide (NaHMDS), THF, 40 °C 66–87%, 5:1 Z:E ratio by 1H NMR. (g) n-BuLi (2.5 m in hexanes), THF, 0 °C→23 °C; ZnCl2; vinyl iodide 9, PdCl2(PPh3)2, 85–95%. (h) Montmorillonite K-10, MeOH, 50 °C. i) Tf2O, 4-dimethylaminopyridine (DMAP), CH2Cl2, 23 °C, 65% yield over two steps. (j) Herrmann–Beller catalyst (17, 10 mol%), Bu4NOAc, DMA, 120 °C (μwaves), 88% (0.12 mmol scale) or 54% (2.0 mmol scale). (k) (R,R)-Mn(salen) catalyst (23, 15 mol %), NaOCl (aq., 0.54 m, pH 11.2), CH2Cl2, 23 °C, 89%, 7.5–10:1 d.r. by 1H NMR analysis. (l) AuCl3 (20 mol%), acetone, 23 °C, 74%, single diastereomer by 1H NMR analysis. (m) MePPh3Br, n-BuLi (2.06m in hexanes), benzene, 23 °C→90 °C, 67%.
References
Wellington, K. D., Cambie, R. C., Rutledge, P. S. & Bergquist, P. R. Chemistry of sponges. 19. Novel bioactive metabolites from Hamigera tarangaensis. J. Nat. Prod. 63, 79–85 (2000).
Nicolaou, K. C., Gray, D. & Tae, J. Total synthesis of hamigerans: Part 1. Development of synthetic technology for the construction of benzannulated polycyclic systems by the intramolecular trapping of photogenerated hydroxy-o-quinodimethanes and synthesis of key building blocks. Angew. Chem. Int. Ed. 40, 3675–3678 (2001).
Nicolaou, K. C., Gray, D. & Tae, J. Total synthesis of hamigerans: Part 2. Implementation of the intramolecular Diels–Alder trapping of photochemically generated hydroxy-o-quinodimethanes; strategy and completion of the synthesis. Angew. Chem. Int. Ed. 40, 3679–3683 (2001).
Mehta, G. & Shinde, H. M. Enantiospecific total synthesis of 6-epi-(–)-hamigeran B. Intramolecular Heck reaction in a sterically constrained environment. Tetrahedron Lett. 44, 7049–7053 (2003).
Clive, D. L. J. & Wang, J. Synthesis of (±)-hamigeran B, (–)-hamigeran B and (±)-1-epi-hamigeran B: use of bulky silyl groups to protect a benzylic carbon–oxygen bond from hydrogenolysis. J. Org. Chem. 69, 2773–2784 (2004).
Sperry, J. B. & Wright, D. L. Synthesis of the hamigeran skeleton through an electro-oxidative coupling reaction. Tetrahedron Lett. 46, 411–414 (2005).
Trost, B. M., Pissot-Soldermann, C. & Chen, I. A short and concise asymmetric synthesis of hamigeran B. Chem. Eur. J. 11, 951–959 (2005).
Taber, D. F. & Tian, W. Synthesis of (–)-hamigeran B. J. Org. Chem. 73, 7560–7564 (2008).
Miesch, L., Welsch, T., Rietsch, V. & Miesch, M. Intramolecular Alknylogous mukaiyama aldol reaction starting from bicyclic alkanones tethered to alkynyl esters: formal total synthesis of (±)-hamigeran B. Chem. Eur. J 15, 4394–4401 (2009).
Cai, Z. & Harmata, M. Studies directed toward the synthesis of hamigeran b: a catalytic oxidative cyclization. Org. Lett. 12, 5668–5670 (2010).
Lau, S. Y. W. Concise and protective group-free synthesis of (±)-hamigeran B and (±)-4-bromohamigeran B. Org. Lett. 13, 347–349 (2011).
Mukherjee, H., McDougal, N. T., Virgil, S. & Stoltz, B. M. A catalytic, asymmetric formal synthesis of (+)-hamigeran B. Org. Lett. 13, 825–827 (2011).
Lin, H. et al. Enantioselective approach to (–)-hamigeran B and (–)-4-bromohamigeran B via Catalytic asymmetric hydrogenation of racemic ketone to assemble the chiral core framework. Org. Lett. 18, 1434–1437 (2016).
Jiang, B., Li, M.-M., Xing, P. & Huang, Z.-G. A concise formal synthesis of (–)-hamigeran B. Org. Lett. 15, 871–873 (2013).
Li, X., Xue, D., Wang, C. & Gao, S. Total synthesis of the hamigerans. Angew. Chem. Int. Ed. 55, 9942–9946 (2016).
Hong, A. Y. et al. Ring-contraction strategy for the practical, scalable, catalytic asymmetric synthesis of versatile γ-quaternary acylcyclopentenes. Angew. Chem. Int. Ed. 50, 2756–2760 (2011).
Mirrington, R. N. & Feutrill, G. I. Orcinol monomethyl ether. Org. Synth. 53, 90–93 (1973).
Stork, G. & Zhao, K. A stereoselective synthesis of (Z)-1-iodo-1-alkenes. Tetrahedron Lett. 30, 2173–2174 (1989).
Balamurugan, R., Kothapalli, R. B. & Thota, G. K. Gold-catalysed activation of epoxides: application in the synthesis of bicyclic ketals. Eur. J. Org. Chem. 2011, 1557–1569 (2011).
Acknowledgements
Dedicated to Prof K. C. Nicolaou for his tremendous scientific contributions to the total synthesis of highly complex and biologically important natural products. We thank NIH-NIGMS (R01GM080269) for supporting this research. DCD would like to thank the NSF (predoctoral research fellowship, No. DGE-1144469) and Caltech for financial support. Dr David VanderVelde (Caltech) is thanked for aid in NMR structural determination. Dr Allen Hong and Dr Scott Virgil are thanked for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dedicated to Professor KC Nicolaou for his tremendous scientific contributions to the total synthesis of highly complex and biologically important natural products.
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Duquette, D., Jensen, T. & Stoltz, B. Progress towards the total synthesis of hamigerans C and D: a direct approach to an elaborated 6-7-5 carbocyclic core. J Antibiot 71, 263–267 (2018). https://doi.org/10.1038/ja.2017.96
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2017.96
This article is cited by
-
A review on various aspects of organic synthesis using Comins’ reagent
Molecular Diversity (2022)
-
Cross-coupling reactions towards the synthesis of natural products
Molecular Diversity (2022)
