
Abstract
Molecular hybridization approach is an emerging tool in drug discovery for designing new pharmacophores with biological activity. A novel, new series of coumarin–benzimidazole hybrids were designed, synthesized and evaluated for their broad spectrum antimicrobial activity. Among all the synthesized molecules, compound (E)-3-(2-1H-benzo[d]imidazol-1-yl)-1-((4-chlorobenzyl)oxy)imino)ethyl)-2H-chromen-2-one showed the most promising broad spectrum antibacterial activity against Pseudomonas aeruginosa, Staphylococcus aureus, Bacillus subtilis and Proteus vulgaris. In addition, it has showed no cytotoxicity and hemolysis at 10 times the MIC concentration. SAR studies indicate that position of the chlorine atom in the hybrid critically determines the antibacterial activity.
Similar content being viewed by others

Introduction
Despite the significant progress in medicine, treating the infectious diseases remains a serious concern to the clinics due to the emergence of multidrug resistance in most of the pathogens.1, 2 According to World Health Organization (WHO), annually 57 million deaths occur globally due to infectious diseases alone and this number is likely to rise in the coming years due to wide spreading of resistance genes across the bacterial species. Globally, multidrug-resistant pathogens especially the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter species) pose a great challenge in clinics as they are capable of ‘escaping’ from the lethal action of existing antimicrobial agents and are able to cause nosocomial infections. In addition to natural acquisition or lateral transfer of antibiotic resistance gene in pathogens, the decrease in new antimicrobials discovery has failed to keep the check on the rise in antibiotic resistance worldwide. It is now accepted in general that lack of new antibiotics will have a serious corollary on the universal public health, security or survival of human race in coming years.
There is an immense and urgent need for a new class of chemical entities with a different mode of action to replace or augment the existing therapeutics. In order to cope up with this problem, various governments as well as agencies such as Infectious Diseases Society of America (IDSA), the European Centre for Disease Prevention and Control (ECDC) and the WHO have started incentives for the research on novel antimicrobial agents against life-threatening infections caused by the multidrug-resistant pathogens.
It is widely reported that the natural compounds can play a major role as lead structures for the development of synthetic molecules.3, 4 Coumarins (2H-chromen-2-one) are naturally occurring compounds with a wide range of biological activities.5, 6, 7, 8, 9, 10, 11 Coumarin-based Novobiocin (1, Figure 1) and Clorobiocin (2, Figure 1) are identified as potent inhibitors of bacterial DNA gyrase.12, 13, 14 Recently, Li et al.15 have reported coumarin-based inhibitors (3, Figure 1) with potent antibacterial activity against multiple ciprofloxacin and methicillin-resistant S. aureus strains. Sahoo and Paidshetty16 reported that the cobalt complexes of 3-aryl-azo-4-hydroxy coumarin analogs have a significant antimicrobial, wound healing and antioxidant properties. Paul et al.17 reported anticancer activity of coumarin–benzimidazole hybrids. Al-Majedy et al.18 has reviewed the previous studies on coumarins with respect to the antimicrobial activity and reported that coumarins analogs exhibited potential antimicrobial activity that is stronger than that of their parent molecules. This work and others have motivated us to design and synthesize new coumarin-based hybrid analogs with enhanced antibacterial activity. Molecular hybridization approach allows integrating two or more pharmacophoric units, possessing different or similar bioactivity, into one molecular scaffold with novel or improved biological activities with respect to the corresponding pharmacophoric units.19, 20, 21 It is often reported that the hybrids offer favorable pharmacokinetics, avoid undesired drawbacks such as side effects and low oral bioavailability, in addition to interesting multiple biological activities, high selectivity. These advantages have made hybrid molecules a rationally attractive source for current drug discovery. Benzimidazole is an important heterocyclic pharmacophore structure with a very strong application in the synthesis of drug-like molecules.22, 23 Literature review indicates that many derivatives of benzimidazole have been successfully developed as clinical drugs and have been widely used in the treatment of various diseases. Recent work indicated that benzimidazoles (4 and 5, Figure 1) possess much potential to inhibit the growth of bacteria.22

Some potent coumarin, benzimidazole antibacterial compounds and general structure of our synthesized coumarin–benzimidazole conjugates. A full color version of this figure is available at The Journal of Antibiotics journal online.
Even though, hybrid approach has given excellent results in the drug discovery program for antimalarial and anticancer agents,24, 25 not much work is done in the area of antimicrobial agents especially coumarin benzimidazoles. These scanty reports regarding the coumarin–benzimidazole hybrids and success stories of hybrid approach lead us to synthesize the novel coumarin–benzimidazole hybrids with special reference to antimicrobial activity.17, 26, 27 We here report the synthesis of the coumarin–benzimidazole hybrids as well as their antimicrobial activity, especially against the ESKAPE pathogens.
Results and Discussion
A simple and efficient method for the preparation of coumarin–benzimidazole conjugates was developed. The synthesized hybrids were evaluated for the antibacterial activity against ESKAPE pathogens as well as cytotoxicity against eukaryotic cells by using the FDA-approved protocols. We observed that the compound 12 ((E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-(((4-chlorobenzyl)oxy)imino)ethyl)-2H-chromen-2-one) has shown a broad spectrum antimicrobial activity. SAR studies showed that in comparison to the others, halogen containing coumarin–benzimidazole conjugates had a broad spectrum activity over the once, which do not have.
Chemistry
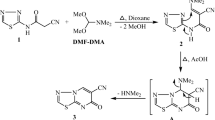
The route followed for the synthesis of coumarin–benzimidazole conjugates is illustrated in Figure 2. The Knoevenagel condensation between salicylaldehyde and ethyl acetoacetate in the presence of catalytic amount of piperidine gave intermediate 8, which on further bromination in dry chloroform resulted in the formation of compound 9.28 Further, the nucleophilic substitution on compound 9 with benzimidazole in acetonitrile at room temperature gave compound 10. Subsequently, condensation of compound 10 with hydroxyl amine hydrogen chloride in ethanol under reflux conditions gave compound 11.29 Finally, the targeted conjugates (12–22) were achieved by etherification of compound 11 with different substituted benzyl halides in DMSO and potassium tert-butoxide as a base. All the new synthesized coumarin–benzimidazole conjugates were characterized using NMR, IR and MS (Supplementary Figure 1). The characterization details were provided in the Supplementary section.

Synthesis of coumarin–benzimidazole conjugates.
Antimicrobial studies
In this present study, 11 novel coumarin–benzimidazole conjugates were screened for their in vitro antibacterial activity against reported Gram-positive and Gram-negative pathogenic bacterial strains by microdilution method as per CLSI guidelines (formerly known as NCLS).30, 31, 32 MIC values were determined by using ampicillin, kanamycin, tetracycline, ciprofloxacin as a reference. The in vitro antibacterial results confirmed that some of the coumarin–benzimidazole conjugates exhibited significant antibacterial activity against two Gram-positive strains such as B. subtilis (MTCC 1789), S. aureus (ATCC 6538P) and three Gram-negative P. aeruginosa (ATCC 25668), Proteus vulgaris (ATCC 29905) and E scherichia coli (MTCC 739) bacterial strains Table 1.
In the antimicrobial assay, out of 11 compounds, compounds 12 and 14 effectively inhibited the growth of the B. subtilis (MTCC 1789) with MIC=0.95 and 6.25 μg ml−1, respectively, whereas reference drug ampicillin had MIC=25 μg ml−1. In the case of P. vulgaris (ATCC 29905), compounds 14, 17, 21 and 22 displayed strong inhibition at MIC 1.56, 3.12, 1.56 and 1.56 μg ml−1 respectively, while ampicillin inhibited at MIC>50 μg ml−1. Compound 12 showed better antibacterial activity against P. aeruginosa (ATCC 25668) with MIC 3.12 μg ml−1 compared to reference drug ampicillin (MIC=25 μg ml−1), tetracycline and kanamycin (MIC>50 μg ml−1). Furthermore, compound 12 showed remarkable activity at MIC 1.56 and 3.12 μg ml−1 against S. aureus (ATCC 6538P) and E. coli (MTCC 739) bacterial strains, respectively. The structure–antimicrobial activity relationship of the synthesized compounds revealed that compounds 14, 17, 21 and 22 inhibited the growth of P. vulgaris ATCC 29905. P. vulgaris belongs to the Enterobacteriaceae family of Gram-negative bacilli and is often implicated in serious sepsis and urinary tract infections in humans, along with ESKAPE species. Plech et al.33 have reported 1,2,4-triazole-ciprofloxacin hybrids as potential antibacterial agents against drug-susceptible and drug-resistant bacteria. CBR-2092, a rifamycin-quinoline hybrid exhibited potent antibacterial activity.34 Coumarin thiosemicarbazone hybrid derivatives were found to have antimicrobial activity against E. coli.35 Furthermore, a series of coumarin-thiazole hybrids were reported as significant antibacterial agents against Gram-positive and Gram-negative bacteria.36
Molecular structural analysis of compounds showed that molecule with a halogen atom has antibacterial activity. We observed that the position and number of halogen atoms in the molecule are also having a significant effect on the activity and host spectrum. Coumarin–benzimidazole conjugate with chlorine atom at para position (compound 12) was found to have highest broad spectrum antimicrobial activity. Interestingly, compound with chlorine atom at meta or ortho position (16 and 22) did not show any antibacterial activity. It is widely known that the molecules biophysical properties can also influence the action spectrum of various therapeutic molecules. EL Hage et al.37 has shown that the number of chlorine atoms and their position on the aromatic ring had a notable effect on the antifungal activity in the bifonazole derivatives. Our results are in consistence with the results obtained with halogenated 1, 2-benzothiazine derivatives compounds, where it was demonstrated that compounds with chlorine or bromine atom in the para position have higher antimicrobial activity over the others.38 Recently it has been demonstrated that the presence of halogen atoms in molecules increases the target binding ability and also stabilizes the ligand–target complex.31 Our observation including others clearly indicates that the position of the chlorine atom is very much important for antibacterial activity.
Hemolysis study
Hemolysis assay is a simple, robust and high throughput assay to detect the toxicity of the compounds. To investigate the toxicity profile, we selected compound 12 as it has shown a broad spectrum antibacterial activity. As these molecules were primarily intended to develop as human therapeutic agents, we tested the toxicity against human erythrocytes. The hemolysis assay results clearly indicate that the selected compounds are not toxic against human erythrocytes even at 10 times the MIC concentration (Figure 3a). Given this, we believe that this molecule is safe and have the potential to become a lead molecule.

Effect of coumarin–benzimidazole conjugates on mammalian cells: (a) Hemolytic effects of compound 12 were investigated. Erythrocytes were incubated with Compound 12 (100 μg ml−1), 2% Triton X-100 (Sigma-Aldrich) served as positive control. The absorbance of hemoglobin release was measured at 540 nm and is expressed as % of Triton X-100-induced hemolysis. (b and c) L929 cells were exposed to Compound 12, coumarin and benzimidazole at indicated concentration for 24 h, and the cell viability was measured by LDH-based kit and MTT assay. Compound 12, coumarin and benzimidazole did not induce any LDH release nor did they affect viability of the cells. 1% Triton X-100 was used as a positive control (yielding 100% LDH release) and buffer as negative presented as controls were present.
Viability assay
Benzimidazoles are structural homolog to purines and their mode of action is by inhibiting the nucleic acid synthesis. As our compound 12 is obtained by the fusion of coumarin and benzimidazole, we tested these molecules in both lactate dehydrogenase (LDH) and MTT assay. We used actively dividing L929 cells (mouse fibroblast cell line) as target cells, LDH assay as cytotoxicity indicator, MTT as an indicator for the cell viability and proliferation. Initially, we exposed the L929 cell lines to the compound 12 at 1 × and 10 × times the MIC concentration for 24 h and then measured the LDH release from dying cells (Figure 3b) and MTT conversion to formazan by metabolically viable cells (Figure 3c). Our results clearly show that the compound 12 is not toxic to L929 cells and does not induce any morphological changes in the structure of the cell (Figure 4). Some of the compounds (14, 16, 17, 18, 21 and 22) synthesized showed detectable antibacterial activity against Gram-positive bacteria B. subtilis and S. aureus. However, their activity was found to be weaker as compared to the reference standard. On the basis of these results, in vitro cytotoxicity test was not conducted for these compounds. Mantu et al.39 reported that benzimidazole and pyridine hybrids have the anticancer activity. Novel series of 1, 2, 3-triazolo-phenanthrene hybrids synthesized by employing Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) showed in vitro cytotoxic against various human cancer cell lines.40

Effect of coumarin–benzimidazole conjugates on mammalian cells morphology. Effects of compound 12, coumarin and benzimidazole on the morphology of L929 cell line was investigated by using phase contrast microscopy. Cells were incubated for 24 h at × 1 (left column) and × 10 (right column), and stained with Giemsa stain before observing under the phase contrast microscope for the morphological changes. A full color version of this figure is available at The Journal of Antibiotics journal online.
Conclusion
In conclusion, we have designed and synthesized a novel series of coumarin–benzimidazole hybrids and evaluated them for their antibacterial activities. The hybrid compound 12 (E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-(((4-chlorobenzyl)oxy)imino)ethyl)-2H-chromen-2-one displayed significant activity against different Gram-positive and Gram-negative bacterial strains. Although the precise target and mode of action remains unknown, given the robust increase in the list of pathogen acquiring the resistance against antibiotics, we believe that these hybrids may serve as promising scaffolds against the treatment of bacterial infections.
Experimental procedures
Analysis and instruments
All reagents were commercially procured from Sigma (Delhi, India) or Spectrochem (Hyderabad, India) India and were used without further purification. Chromatography was carried on silica gel (60–120 and 100–200 mesh). All reactions were monitored by TLC; silica gel plates with fluorescence F254 were used. Melting points were uncorrected. The 1H NMR, and 13C NMR spectra were determined on 200, 300, 50, 75, MHz, respectively, using CDCl3 and DMSO- d6 as solvents and tetramethylsilane (TMS) as an internal standard. All chemical shifts were given in p.p.m. IR spectra were recorded on in the range of 500–4000 cm−1 and multiplicity (s=singlet, brs=broad singlet, d=doublet, brd=broad doublet, dd==double doublet, t=triplet, q==quartet, m=multiplet).
Synthesis of 3-(1H-benzo[d]imidazole-1-carbonyl)-2H-chromen-2-one (10)
The intermediates 8 and 9 were synthesized according to the literature procedure with 92% and 88% yields, respectively.28 Further, the brominated compound 9 (2.0 g, 7.5 mmol) was subjected to reaction with benzimidazole (890 mg, 7.5 mmol) in the presence of 30 ml of acetonitrile at room temperature. The reaction mixture was kept stirring for 3 h. Upon complete consumption of reactants, the reaction was quenched with saturated aq.NaHCO3 solutions and extracted with 10 ml of ethyl acetate three times. The combined solvent fractions were dried over anhydrous Na2SO4 and evaporated under reduced pressure. The resulting crude product thus obtained was purified by column chromatography (using 100–200 mesh silica gel and MeOH/CHCl3 as eluent). Subsequently pure compound 10 was obtained. Yellow solid, yield: 71%, that is, 631 mg; mp 150–152 °C; IR (KBr, cm−1): 3014, 2887, 2359, 1743, 1657, 1402, 1199, 1087; 1H NMR (400 MHz, DMSO-d6,): δ 8.84 (s, 1H), 8.19 (s, 1H), 8.04 (d, J=7.8 Hz, 1H), 7.85–7.80 (m, 1H), 7.68 (bs, 1H), 7.57 (d, J=8.4 Hz, 1H), 7.50–7.45 (m, 2H), 7.23–7.21 (m, 2H), 5.89 (s, 2H); ESI–MS (m/z): 305 [M+H]+.
Synthesis of (E)-3-((1H-benzo[d]imidazol-1-yl) (hydroxyimino) methyl)-2H-chromen-2-one (11)
The reaction of compound 10 (2.5 g, 8.2 mmol) was carried out with hydroxyl amine hydrochloride (655 mg, 9.8 mmol) in the presence of 30 ml of ethanol under refluxing conditions at 70–80 °C for 4 h. On completion of the reaction (monitored by TLC), the excess solvent was removed under reduced pressure. The resulted crude product was purified by column chromatography (using 100–200 mesh silica gel and MeOH/CHCl3 as eluent) to obtain pure compound 11. Yellow solid, yield: 65%, that is, 1.625 g ; mp 178–179 °C; IR (KBr, cm−1): 3412, 3019, 2889, 2399, 1652, 1405, 1215, 1095; 1H NMR (400 MHz, DMSO-d6): δ 12.35 (s, 1H), 8.10 (s, 1H), 8.05 (s, 1H), 7.68 (d, J=7.4 Hz, 1H), 7.61–7.54 (m, 2H), 7.43 (d, J=7.8 Hz, 1H), 7.36 (d, J=8.0 Hz, 1H), 7.32–7.29 (m, 1H), 7.24–7.20 (m, 1H), 7.16–7.12 (m, 1H), 5.53 (s, 2H); ESI–MS (m/z): 320 [M+H]+.
Synthesis of (E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-(((4-chlorobenzyl)oxy)imino)ethyl) -2H-chromen-2-one (12)
Compound 11 (250 mg, 0.78 mmol) was dissolved in 4 ml of DMSO and then potassium tert-butoxide (130 mg, 1.1 mmol) was added to it. The resulting reaction mixture was kept stirring at room temperature. After 5 min 4-chlorobenzyl chloride (125 mg, 0.78 mmol) was added and the reaction mixture was kept stirring at room temperature for about 1h. After completion of reaction (monitored by TLC), the crude product was extracted with ethyl acetate. The combined organic layers were dried on anhydrous sodium sulfate, concentrated under reduced pressure and purified by column chromatography (100–200 mesh neutral silica gel) to afford the pure compound 12. White solid, yield: 67%, that is, 167.5 mg; mp 130–132 °C; IR (KBr, cm−1): 3021, 2401, 1718, 1616, 1494, 1427, 1215, 1025; 1H NMR (400 MHz, CDCl3): δ 7.97 (s, 1H), 7.71 (d, J=7.6 Hz, 1H), 7.67 (s, 1H), 7.53–7.49 (m, 1H), 7.41–7.35 (m, 5H), 7.28 (d, J=8.3 Hz, 1H), 7.24–7.19 (m, 4H), 5.66 (s, 2H), 5.31 (s, 2H); 13C NMR (100 MHz, CDCl3): δ 160.0, 154.1, 152.1, 144.0, 143.7, 143.2, 135.1, 134.6, 133.6, 133.0, 130.1, 129.0, 128.7, 124.9, 123.3, 122.5, 121.1, 120.4, 118.4, 116.7, 109.4, 76.8, 39.7; HRMS (ESI) calcd for C25H18ClN3O3 [M + H]+ 444.1115 found 444.1067. The compounds 13–22 were prepared by similar procedure as described for 12 using appropriately substituted benzyl halides.
(E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-(((4-fluorobenzyl)oxy)imino)ethyl)-2H-chromen-2-one (13)
White solid, yield: 71%, that is, 177.5 mg; mp 168–170 °C; IR (KBr, cm−1): 3023, 2415, 1720, 1608, 1500, 1370, 1216, 1019; 1H NMR (400 MHz, CDCl3): δ 7.95 (s, 1H), 7.71–7.67 (m, 2H), 7.53–7.49 (m, 1H), 7.43–7.40 (m, 2H), 7.36 (d, J=7.4 Hz, 1H), 7.28–7.25 (m, 1H), 7.23–7.18 (m, 4H), 7.13–7.09 (m, 2H), 5.65 (s, 2H), 5.31 (s, 2H); 13C NMR (100 MHz, CDCl3,): δ 164.2, 161.7, 160.0, 154.1, 152.0, 144.1, 143.7, 133.7, 133.0, 132.9, 132.4, 130.8, 130.7, 128.7, 124.9, 123.2, 122.4, 121.2, 120.4, 118.5, 116.7, 115.8, 115.6, 109.4, 76.8, 39.6; HRMS (ESI) calcd for C25H18FN3O3 [M + H]+ 428.1410 found 428.1399.
(E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-(((4-bromobenzyl)oxy)imino)ethyl)-2H-chromen-2-one (14)
White solid, yield: 63%, that is, 157.6 mg; mp 124–126 °C; IR (KBr, cm−1): 3019, 2400, 1721, 1608, 1490, 1405, 1215, 1026; 1H NMR (400 MHz, CDCl3): δ 7.94 (s, 1H), 7.70 (d, J=7.3 Hz, 1H), 7.60 (s, 1H), 7.55 (d, J=8.3 Hz, 2H), 7.53–7.49 (m, 1H), 7.36 (dd, J=7.7, 1.2 Hz, 1H), 7.30 (d, J=8.2 Hz, 2H), 7.27 (d, J=8.6 Hz, 1H), 7.23–7.20 (m, 2H), 7.19–7.17 (m, 2H), 5.65 (s, 2H), 5.29 (s, 2H); 13C NMR (100 MHz, CDCl3): δ 160.0, 154.2, 152.3, 144.1, 143.7, 135.6, 133.8, 133.0, 131.9, 130.4, 128.7, 124.9, 123.2, 122.7, 122.4, 121.2, 120.5, 118.5, 116.7, 109.4, 76.7, 39.7; HRMS (ESI) calcd for C25H18BrN3O3 [M + H]+ 488.0610 found 488.0584.
(E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-(3-(trifluoromethyl)benzyloxyimino)ethyl)-2H-chromen-2-one (15)
Whitish yellow solid, yield: 71%, that is, 177.2 mg; mp 146–148 °C; IR (KBr, cm−1): 3001, 2517, 1722, 1611, 1473, 1384, 1250, 1026; 1H NMR (400 MHz, CDCl3): δ 7.94 (s, 1H), 7.75 (s, 1H), 7.70–7.66 (m, 2H), 7.64–7.62 (m, 2H), 7.58 (d, J=7.6 Hz, 1H), 7.51–7.49 (m, 1H), 7.34 (dd, J==7.7 Hz, J=1.1 Hz, 1H), 7.28–7.26 (m, 1H), 7.23–7.18 (m, 2H), 7.16–7.15 (m, 2H), 5.67 (s, 2H), 5.40 (s, 2H); 13C NMR (75 MHz, CDCl3): δ 160.0, 154.2, 152.6, 144.1, 143.9, 143.5, 137.7, 133.7, 133.0, 132.0, 131.4, 130.9, 129.4, 128.7, 125.9, 125.5, 125.0, 123.3, 122.5, 121.0, 120.5, 118.4, 116.7, 109.2, 76.7, 39.7; HRMS (ESI) calcd for C26H18F3N3O3 [M + H]+ 478.1379 found 478.1360.
(E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-(((3-chlorobenzyl)oxy)imino)ethyl)-2H-chromen-2-one (16)
White solid, yield: 70%, that is, 170.0 mg; mp 148–150 °C; IR (KBr, cm−1): 3019, 2413, 1721, 1627, 1494, 1431, 1215, 1028; 1H NMR (400 MHz, CDCl3): δ 7.96 (s, 1H), 7.70–7.69 (m, 1H), 7.66 (s, 1H), 7.53–7.49 (m, 1H), 7.46 (s, 1H), 7.38–7.36 (m, 2H), 7.34 (s, 1H), 7.32–7.31 (m, 1H), 7.28–7.26 (m, 1H), 7.23–7.20 (m, 4H), 5.68 (s, 2H), 5.32 (s, 2H); 13C NMR (100 MHz, CDCl3): δ 159.9, 154.1, 152.3, 144.0, 143.8, 143.3, 138.6, 134.6, 133.7, 133.0, 130.1, 128.7, 126.7, 124.9, 123.4, 122.5, 121.0, 120.4, 118.4, 116.7, 109.4, 76.7, 39.7; HRMS (ESI) calcd for C25H18ClN3O3 [M + H]+ 444.1115 found 444. 1067.
(E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-((benzyloxy)imino)ethyl)-2H-chromen-2-one (17)
White solid, yield: 61%, that is, 152.2 mg; mp 144–146 °C; IR (KBr, cm−1): 3020, 2400, 1717, 1617, 1521, 1424, 1215, 1039; 1H NMR (400 MHz, CDCl3,): δ 7.94 (s, 1H), 7.70–7.67 (m, 2H), 7.52–7.48 (m, 1H), 7.47–7.44 (m, 4H), 7.42–7.40 (m, 1H), 7.36–7.34 (m, 1H), 7.28–7.25 (m, 1H), 7.22–7.15 (m, 4H), 5.67 (s, 2H), 5.37 (s, 2H); 13C NMR (100 MHz, CDCl3,): δ 160.0, 154.1, 151.9, 144.1, 143.7, 143.5, 136.6, 133.7, 132.9, 128.8, 124.8, 123.2, 122.3, 121.2, 120.4, 118.5, 116.7, 109.5, 76.7, 39.6; HRMS (ESI) calcd for C25H19N3O3 [M + H]+ 410.1505 found 410.1480.
(E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-(((3-nitrobenzyl)oxy)imino)ethyl)-2H-chromen-2-one (18)
Yellow solid, yield: 72%, that is, 180 mg; mp 160–162 °C; IR (KBr, cm−1): 3021, 2425, 1719, 1611, 1492, 1456, 1219, 1023; 1H NMR (400 MHz, CDCl3,): δ 8.35 (s, 1H), 8.25 (d, J =7.3 Hz, 1H), 8.03 (bs, 1H), 7.71 (bs, 3H), 7.63–7.59 (m, 1H), 7.55–7.51 (m, 1H), 7.39 (d, J==7.1 Hz, 1H), 7.29–7.21 (m, 5H), 5.68 (s, 2H), 5.43 (s, 2H); 13C NMR (100 MHz, CDCl3,): δ 159.9, 154.1, 152.8, 148.5, 144.0, 142.8, 138.7, 134.5, 133.1, 129.9, 128.8, 125.0, 123.5, 122.7, 120.8, 120.3, 118.4, 116.7, 109.4, 76.1, 39.9; HRMS (ESI) calcd for C25H18N4O5 [M + H]+ 455.1355 found 455.1304.
(E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-(3-methylbenzyloxyimino)ethyl)-2H-chromen-2-one (19)
Yellow solid, yield: 75%, that is, 187 mg; mp 157–159 °C; IR (KBr, cm−1): 2979, 2410, 1712, 1623, 1479, 1379, 1235, 1021; 1H NMR (400 MHz, CDCl3,): δ 7.94 (s, 1H), 7.70–7.68 (m, 2H), 7.52–7.48 (m, 1H), 7.35–7.31 (m, 2H), 7.27–7.26 (m, 3H), 7.22–7.16 (m, 5H), 5.67 (s, 2H), 5.33 (s, 2H), 2.41 (s, 3H); 13C NMR (75 MHz, CDCl3,): δ 160.0, 154.1, 151.8, 144.1, 143.7, 143.4, 138.4, 136.5, 133.7, 132.8, 129.5, 129.4, 128.7, 125.8, 124.8, 123.2, 122.4, 121.2, 120.3, 118.5, 116.7, 109.5, 77.8, 39.6, 21.5; HRMS (ESI) calcd for C26H21N3O3 [M + H]+ 424.1661 found 424.1649.
(E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-(((4-methoxybenzyl)oxy)imino)ethyl)-2H-chromen-2-one (20)
White solid, yield: 73%, that is, 182 mg; mp 126–128 °C; IR (KBr, cm−1): 3019, 2400, 1721, 1611, 1495, 1406, 1215, 1038; 1H NMR (400 MHz, CDCl3,): δ 7.93 (s, 1H), 7.69–7.68 (m, 2H), 7.52–7.48 (m, 1H), 7.40 (d, J=8.5 Hz, 2H), 7.35 (d, J=7.7 Hz, 1H), 7.28–7.25 (m, 1H), 7.23–7.15 (m, 4H), 6.96 (d, J=8.6 Hz, 2H), 5.65 (s, 2H), 5.29 (s, 2H), 3.85 (s, 3H); 13C NMR (100 MHz, CDCl3,): δ 160.0, 154.1, 151.6, 144.1, 143.6, 143.5, 133.7, 132.8, 130.6, 128.7, 124.8, 123.2, 122.3, 121.3, 120.3, 118.5, 116.7, 114.1, 109.5, 77.4, 55.4, 39.5; HRMS (ESI) calcd for C26H21N3O4 [M + H]+ 440.1610 found 440.1603.
(E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-(((2,4-difluorobenzyl)oxy)imino)ethyl)-2H-chromen-2-one (21)
White solid, yield: 68%, that is, 170 mg; mp 140–142 °C; IR (KBr, cm−1): 3020, 2411, 1719, 1615, 1499, 1457, 1277, 1019; 1H NMR (400 MHz, CDCl3): δ 7.94 (s, 1H), 7.70–7.67 (m, 2H), 7.53–7.49 (m, 1H), 7.45–7.39 (m, 1H), 7.36 (d, J=6.8 Hz, 1H), 7.27 (d, J=8.6 Hz, 1H), 7.24–7.18 (m, 4H), 6.95–6.89 (m, 2H), 5.67 (s, 2H), 5.38 (s, 2H); 13C NMR (100 MHz, CDCl3): δ 165.0, 163.2, 161.7, 160.0, 154.2, 152.3, 144.0, 143.7, 143.5, 133.8, 133.0, 132.4, 128.7, 124.9, 123.2, 122.4, 121.1, 120.5, 118.5, 116.7, 109.4, 104.3, 70.8, 39.5; HRMS (ESI) calcd for C25H17F2N3O3 [M + H]+ 446.1316 found 446.1293.
(E)-3-(2-(1H-benzo[d]imidazol-1-yl)-1-(((2,5-dichlorobenzyl)oxy)imino)ethyl)-2H-chromen-2-one (22)
Yellow solid, yield: 71%, that is, 177 mg; mp 136–138 °C; IR (KBr, cm−1): 2949, 2417, 1719, 1610, 1459, 1364, 1233, 1024; 1H NMR (400 MHz, CDCl3): δ 8.42 (s, 1H), 7.83–7.82 (m, 2H), 7.57–7.53 (m, 1H), 7.43–7.41 (m, 2H), 7.40–7.36 (m, 2H), 7.31–7.29 (m, 4H), 7.26–7.23 (m, 1H), 5.75 (s, 2H), 5.41 (s, 2H); 13C NMR (100 MHz, CDCl3): δ 159.9, 154.1, 151.4, 144.3, 142.6, 138.5, 135.6, 133.3, 132.9, 132.3, 131.0, 130.6, 130.0, 128.9, 125.1, 124.7, 124.2, 120.4, 118.9, 118.4, 116.7, 110.2, 76.4, 40.4; HRMS (ESI) calcd for C25H17Cl2N3O3 [M + H]+ 478.0725 found 478.0718.
Ethical statement
This study has the prior approval from the CSIR-CDRI institutional human ethics committee at CDRI, Lucknow (CDRI/IEC/2014/A1).
Bacterial strains
All the bacterial strains were obtained from the MTCC, India or ATCC, USA and were routinely grown in Nutrient agar medium (NAM; Difco/Becton & Dickinson, Franklin Lakes, NJ, USA). Just before the experiment, a loop full of bacteria was inoculated in Mueller Hinton (MH) broth and incubated at 37 °C and 5% CO2 for overnight to get the starter culture.
MIC assay
MIC assay for the test compounds was carried out by the microtitre broth dilution method as prescribed by CLSI (previously NCCLS).20, 21 For determination of MIC, test compounds were diluted from a stock solution (10 mg ml−1) such that the final concentration ranges between 50 and 0.39 μg ml−1 in different wells. Bacteria grown in 5% MH broth for 12–18 h were rinsed twice with sterile phosphate-buffered saline and diluted in MH broth to a concentration of ~5 × 105 CFU per ml. About 90 μl of bacterial suspension in MH medium was added to each well containing the test compound and incubated at 37 °C for 16 h. A positive control for growth containing no test compound was included in each assay. MIC was taken as the concentration where the growth inhibition observed was >95%. For each test compound, MIC determinations were carried independently twice using triplicate samples each time.
Hemolysis assay
Hemolysis assay was done using human blood, collected by venipuncture technique in EDTA (2 mg ml−1) containing vacationers from the volunteers. The collected blood was centrifuged at 800 g for 10 min, followed by the removal of plasma and buffy coat from the supernatant. The erythrocytes containing pellet was washed three times in phosphate-buffered saline, pH 7.4 and resuspended in the same solution to get a 5% suspension. Later erythrocytes suspension was incubated with end-over-end rotation for 1 h at 37 °C in the presence of test compounds at (100 μg ml−1). After the incubation, samples were centrifuged at 800 g for 10 min and the supernatant was transferred to a 96-well plate. Hemoglobin release was measured by reading the absorbance at 540 nm and is expressed as a percentage of Triton X-100-induced hemolysis. For positive control, 2% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA) was used. The results given represent mean values from triplicate measurements.

MTT assay
The viability of L929 cell lines exposed to test compound was analyzed by using MTT assay. About 5 mg of MTT (3-(4,5-dimethylthiazolyl)-2,5-diphenyl-tetrazoliumbromide; Sigma-Aldrich, Cat No M5655) was dissolved in 1 ml of phosphate-buffered saline and filter sterilized in brown color eppendorf tubes before storing at −20 °C. Two hundred μl of L929 cell line (2.5 × 105 cells per ml) was plated in 96-well plates and grown in DMEM (Sigma, Cat. No D5525) supplemented with 2 mm Glutamine, 0.2% sodium bicarbonate and 10% fetal bovine serum for 2 h before adding the test compounds at 1 × and 10 × times of MIC concentration incubated for 18–24 h at 37 °C and 5% CO2. After the incubation, the 20 μl of MTT solution (5 mg ml−1) was added and further incubated for 3–4 h at 37 °C and 5% CO2. Medium containing the MTT was removed and 100 μl of DMSO was added to dissolve the Formazan formed from MTT by the enzymes associated with metabolic activity. The plates were gently swirled for 10 min at room temperature to dissolve the precipitate and the absorbance was monitored at 550 nm. The results presented here are mean values from the independent triplicate experiment.
Lactate dehydrogenase assay
LDH assay was done to show that test compounds are safe and do not induce any cell death. L929 cell lines (2.5 × 105 cells per ml) were grown in 96-well plates to confluency and test compounds at 1 × and 10 × times of MIC concentration were added in triplicates to respective labeled wells of the plate, followed by incubation for 18–24 h at 37 °C and 5% CO2. After incubation, 100 μl of the medium was transferred into fresh 96-well plates and the LDH release from the cells was quantified by using Cytotoxicity Detection Kit (Roche, Indianapolis, IN, USA, Cytotoxicity Detection Kit, Cat # 11 644 793 001) as per manufacture instructions. The mean values represented here are from three independent experiments and are given as fractional LDH release compared to the positive control consisting of 1% Triton X-100 (yielding 100% LDH release).23
References
Khan, S. N. & Khan, A. U. Breaking the spell: combating multidrug resistant 'Superbugs'. Front. Microbiol. 7, 174 (2016).
Tommasi, R., Brown, D. G., Walkup, G. K., Manchester, J. I. & Miller, A. A. ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug Discov. 14, 529–542 (2015).
Newman, D. J. & Cragg, G. M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661 (2016).
Vuorelaa, P. et al. Natural products in the process of finding new drug candidates. Curr. Med. Chem. 11, 1375–1389 (2004).
Appendino, G. et al. Antimycobacterial coumarins from the sardinian giant fennel (Ferula communis). J. Nat. Prod. 67, 2108–2110 (2004).
Binda, C. et al. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: safinamide and coumarin analogs. J. Med. Chem. 50, 5848–5852 (2007).
Kontogiorgis, C. A. & Hadjipavlou-Litina, D. J. Synthesis and biological evaluation of novel coumarin derivatives with a 7-azomethine linkage. Bioorg. Med. Chem. Lett. 14, 611–614 (2004).
Ma, T. et al. Chemical library and structure-activity relationships of 11-demethyl-12-oxo calanolide A analogues as anti-HIV-1 agents. J. Med. Chem. 51, 1432–1446 (2008).
Sashidhara, K. V. et al. Molecular iodine catalysed one-pot synthesis of chromeno[4,3-b]quinolin-6-ones under microwave irradiation. Green Chem. 17, 3766–3770 (2015).
Shi, Y. & Zhou, C. H. Synthesis and evaluation of a class of new coumarin triazole derivatives as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 21, 956–960 (2011).
Tambo-ong, A., Chopra, S., Glaser, B. T., Matsuyama, K., Tran, T. & Madrid, P. B. Mannich reaction derivatives of novobiocin with modulated physiochemical properties and their antibacterial activities. Bioorg. Med. Chem. Lett. 21, 5697–5700 (2011).
Laurin, P. et al. Synthesis and in vitroevaluation of novel highly potent coumarin inhibitors of gyrase B. Bioorg. Med. Chem. Lett. 9, 2079–2084 (1999).
Schimana, J. et al. Simocyclinones, novel cytostatic angucyclinone antibiotics produced by Streptomyces antibioticus Tu 6040. I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 53, 779–787 (2000).
Watt, P. M. & Hickson, I. D. Structure and function of type II DNA topoisomerases. Biochem. J. 303 (Pt 3), 681–695 (1994).
Li, B. et al. Coumarin-based inhibitors of Bacillus anthracis and Staphylococcus aureus replicative DNA helicase: chemical optimization, biological evaluation, and antibacterial activities. J. Med. Chem. 55, 10896–10908 (2012).
Sahoo, J. & Paidesetty, S. K. Antimicrobial activity of novel synthesized coumarin based transitional metal complexes. J. Taibah Univ. Med. Sci. 12, 115–124 (2017).
Paul, K., Bindal, S. & Luxami, V. Synthesis of new conjugated coumarin-benzimidazole hybrids and their anticancer activity. Bioorg. Med. Chem. Lett. 23, 3667–3672 (2013).
Al-Majedy, Y. K., Kadhum, A. A. H., Al-Amiery, A. A. & Abu Bakar Mohamad, A. B. Coumarins: the antimicrobial agents. Sys. Rev. Pharm. 8, 62–70 (2017).
Sashidhara, K. V. et al. Designing, synthesis of selective and high-affinity chalcone-benzothiazole hybrids as Brugia malayi thymidylate kinase inhibitors: in vitro validation and docking studies. Eur. J. Med. Chem. 103, 418–428 (2015).
Sashidhara, K. V. et al. Design, synthesis and anticancer activity of dihydropyrimidinone-semicarbazone hybrids as potential human DNA ligase 1 inhibitors. Med. Chem. Commun. 7, 2349–2363 (2016).
Viegas-Junior, C., Danuello, A., da Silva Bolzani, V., Barreiro, E. J. & Fraga, C. A. Molecular hybridization: a useful tool in the design of new drug prototypes. Curr. Med. Chem. 14, 1829–1852 (2007).
Narasimhan, B., Sharma, D. & Kumar, P. Benzimidazole: a medicinally important heterocyclic moiety. Med. Chem. Res. 21, 269–283 (2012).
Zhang, D. et al.. Design, synthesis and antibacterial activity of novel actinonin derivatives containing benzimidazole heterocycles. Eur. J. Med. Chem. 44, 2202–2210 (2009).
Walsh, J. J. & Bell, A. Hybrid drugs for malaria. Curr. Pharm. Des. 15, 2970–2985 (2009).
Teiten, M. H., Dicato, M. & Diederich, M. Hybrid curcumin compounds: a new strategy for cancer treatment. Molecules 19, 20839–20863 (2014).
Arora, R. K., Kaur, N., Bansal, Y. & Bansal, G. Novel coumarin-benzimidazole derivatives as antioxidants and safer anti-inflammatory agents. Acta Pharm. Sin. B 4, 368–375 (2014).
Holiyachi, M. et al. Design, synthesis and structure-activity relationship study of coumarin benzimidazole hybrid as potent antibacterial and anticancer agents. ChemistrySelect 1, 4638–4644 (2016).
Vijesh, A. M., Isloor, A. M., Prabhu, V., Ahmad, S. & Malladi, S. Synthesis, characterization and anti-microbial studies of some novel 2,4-disubstituted thiazoles. Eur. J. Med. Chem. 45, 5460–5464 (2010).
Soltani Rad, M. N., Khalafi-Nezhad, A. & Behrouz, S. Design and synthesis of some novel oxiconazole-like carboacyclic nucleoside analogues, as potential chemotherapeutic agents. Helv. Chim. Acta 92, 1760–1774 (2009).
Hanel, H. & Raether, W. A more sophisticated method of determining the fungicidal effect of water-insoluble preparations with a cell harvester, using miconazole as an example. Mycoses 31, 148–154 (1988).
Hernandes, M. Z., Cavalcanti, S. M., Moreira, D. R., de Azevedo Junior, W. F. & Leite, A. C. Halogen atoms in the modern medicinal chemistry: hints for the drug design. Curr. Drug Targets 11, 303–314 (2010).
Liaras, K., Geronikaki, A., Glamoclija, J., Ciric, A. & Sokovic, M. Thiazole-based chalcones as potent antimicrobial agents. Synthesis and biological evaluation. Bioorg. Med. Chem. 19, 3135–3140 (2011).
Plech, T., Wujec, M., Kosikowska, U., Malm, A., Rajtar, B. & Polz-Dacewicz, M. Synthesis and in vitro activity of 1,2,4-triazole-ciprofloxacin hybrids against drug-susceptible and drug-resistant bacteria. Eur. J. Med. Chem. 60, 128–134 (2013).
Robertson, G. T. et al. In vitro evaluation of CBR-2092, a novel rifamycin-quinolone hybrid antibiotic: studies of the mode of action in Staphylococcus aureus. Antimicrob. Agents Chemother. 52, 2313–2323 (2008).
Vekariya, R. H., Patel, K. D., Rajani, D. P., Rajani, S. D. & Patel, H. D. A one pot, three component synthesis of coumarin hybrid thiosemicarbazone derivatives and their antimicrobial evolution. J. Assoc. Arab Univ. Basic Appl. Sci. 23, 10–15 (2017).
Aggarwal, R., Kumar, S., Kaushik, P., Kaushik, D. & Gupta, G. K. Synthesis and pharmacological evaluation of some novel 2-(5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazol-1-yl)-4-(coumarin-3-yl)thiazole s. Eur. J. Med. Chem. 62, 508–514 (2013).
El Hage, S., Lajoie, B., Feuillolay, C., Roques, C. & Baziard, G. Synthesis, antibacterial and antifungal activities of bifonazole derivatives. Arch. Pharm. 344, 402–410 (2011).
Patel, C. et al. Synthesis and antimicrobial activity of 1,2-benzothiazine derivatives. Molecules 21, 861 (2016).
Mantu, D., Antoci, V., Moldoveanu, C., Zbancioc, G. & Mangalagiu, I. I. Hybrid imidazole (benzimidazole)/pyridine (quinoline) derivatives and evaluation of their anticancer and antimycobacterial activity. J. Enzyme Inhib. Med. Chem. 31 (sup2), 96–103 (2016).
Kumar, N. P., Nekkanti, S., Sujana Kumari, S., Sharma, P. & Shankaraiah, N. Design and synthesis of 1,2,3-triazolo-phenanthrene hybrids as cytotoxic agents. Bioorg. Med. Chem. Lett. 27, 2369–2376 (2017).
Acknowledgements
We are grateful to the Director, CDRI, Lucknow, India for constant encouragement in drug development program, Dr SP Singh for technical support, SAIF for NMR, IR and Mass spectral data. LRS, ASR, AS and GRP are thankful to CSIR, New Delhi, India for the financial support. SR is thankful to ICMR, New Delhi, India for junior research fellowship. CSIR-CDRI communication number: 302/2015/KVS.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Singh, L., Avula, S., Raj, S. et al. Coumarin–benzimidazole hybrids as a potent antimicrobial agent: synthesis and biological elevation. J Antibiot 70, 954–961 (2017). https://doi.org/10.1038/ja.2017.70
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2017.70
This article is cited by
-
In vitro antimicrobial evaluation and in silico studies of coumarin derivatives tagged with pyrano-pyridine and pyrano-pyrimidine moieties as DNA gyrase inhibitors
Molecular Diversity (2022)
-
Synthesis and evaluation of antimicrobial, antitubercular and anticancer activities of 2-(1-benzoyl-1H-benzo[d]imidazol-2-ylthio)-N-substituted acetamides
Chemistry Central Journal (2018)
-
Coumarin: a novel player in microbial quorum sensing and biofilm formation inhibition
Applied Microbiology and Biotechnology (2018)
