
Abstract
Microbial metabolites have attracted increasing interest as a source of therapeutics and as probes for biological mechanisms. New microbial metabolites and derivatives targeted at inflammation and bone disease therapy have been identified by focusing on prostaglandin release, osteoblast differentiation and immune cell functions. These modulators of inflammatory processes and bone disease contribute to our understanding of biological mechanisms and support identification of the therapeutic potential of drug lead candidates. The present review describes recent advances in the chemistry and analysis of inhibitors of prostaglandin release or other functional molecules of immune cells, as well as inducers of osteoblast differentiation, including biological and pharmacological activities.
Similar content being viewed by others

Introduction
Professor Hamao Umezawa directed fundamental studies underpinning the development of antibiotics and their derivatives for use in the treatment of infectious disease and cancer during 1960s–1980s. Considering the importance of inflammatory processes as drug targets, Umezawa and Aoyagi commenced exploratory research to identify low-MW enzyme inhibitors from microbial metabolites. Their discovery of numerous protease inhibitors has permitted analysis of disease mechanisms and therapies for inflammation.1 Of the proteases involved in inflammatory processes, serine–cysteine proteases, plasmin, chymotrypsin, elastase, prolyl endopeptidase and thiol protease have crucial roles in the coagulation, fibrinolytic and kinin systems. Leupeptin,2 chymostatin3 and poststatin4 were identified as inhibitors of plasmin, chymotrypsin and prolyl endopeptidase, respectively by, Aoyagi et al. In addition, elasnin was discovered as an inhibitor of elastase by Omura et al.5
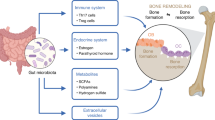
Research on inflammation has since expanded to include inflammatory bone disease. Dysregulated inflammation leads to increased bone resorption and suppressed bone formation. Thus, the inhibition of bone resorption is a therapeutic target for which inhibitors have been screened. For example, ICM0201 isolated from Cunninghamella sp. F-1490 was discovered as an inhibitor of osteoclastogenesis.6, 7
Our recent efforts have directed mechanic study or drug discovery for inflammation and bone disease, focusing on the relationship between inflammation and bone disease, focusing on the following fields: prostaglandin release inhibitors during inflammation; regulating of immune cells during inflammation; and bone repair during bone disease.
Prostaglandin release inhibitors during inflammation
In addition to playing an important role in maintaining physiological homeostasis, prostaglandins have been implicated in several diseases such as inflammatory disorders, sensory neuron-related disorders and cancer. Prostaglandin release inhibitors have the potential to ameliorate inflammation. Thus, a series of prostaglandin release inhibitors are overviewed. Notably, isolation of pronqodine A8 and sacchathridine A9 (two prostaglandin E2 (PGE2) release inhibitors) from microbial metabolites and their biological evaluation are described.
Regulation of immune cells during inflammation
Rheumatoid arthritis,10 a chronic systemic disease characterized by inflammatory erosive synovitis, elicits marked neovascularization, inflammatory cell infiltration and synovial hyperplasia. The infiltration of cells, predominantly monocytes and macrophages into arthritic joints, is observed. Moreover, vascular endothelial growth factor receptor-1 (VEGFR-1; Flt-1) is functionally expressed in both monocytes and macrophages, which have a crucial role in promoting inflammation.11, 12, 13, 14, 15, 16 During screening of microbial metabolites to identify novel inhibitors of VEGFR-1 (Flt-1) tyrosine kinase, vegfrecine17 was isolated from the culture broth of Streptomyces sp. MK931-CF8.
Recently, heparanase (endo-β-d-glucuronidase) present within immune cells has been focused on as an inflammatory modulator. Though heparanase is highly relevant for tumor progression and metastasis, the role of heparanase in inflammatory disorders has also attracted much attention in the past decade because a significant increase in the expression and enzymatic activity of heparanase has been reported in numerous inflammatory conditions, typically associated with degradation of heparan sulfate and extensive remodeling of the extracellular matrix.18, 19 Heparanase inhibitors exhibiting anti-inflammatory effects were overviewed, and inflammatory mechanisms associated with heparanase were studied and elucidated using heparastatin (SF4),20 a synthetic derivative of microbial metabolite siastatin B.21
Bone repair during bone disease
Dysregulation of bone loss and subsequent repair is germane to many orthopedic conditions, including inflammatory arthritis, osteoporosis, osteonecrosis, metabolic bone disease, tumors and periprosthetic particle-associated osteolysis. Bone diseases, such as osteoporosis,22 are caused by dysregulated bone remodeling characterized by the sequential tethering of bone formation by osteoblasts and bone resorption by osteoclasts. Understanding the molecular mechanisms for underlying regulation of osteoblast differentiation is of great interest for developing therapeutics for bone disease and bone regenerative medicine. A series of synthetic and natural inducers of osteoblast differentiation have been summarized. Compounds capable of inducing osteoblast differentiation in C3H10T1/2 cells were screened from libraries of microbial fermentation broths using alkaline phosphatase (ALP) expression. Isolation and biological evaluation of decalpenic acid (DPA),23 discovered within the fermentation broth of Penicillium verruculosum CR37010, have also been described.
This review describes recent progress for studies in investigating the chemistry, biochemistry and pharmacology of pronqodine A, sacchathridine A, DPA, vegfrecine and heparastatin (SF4).
Prostaglandin release inhibitors during inflammation
Prostaglandins are bioactive lipid mediators. Arachidonic acid, key for prostaglandin biosynthesis, is enzymatically converted to various kinds of prostaglandins via cyclooxygenase (COX) and prostaglandin synthase (Figure 1). Each prostaglandin binds to its specific G protein-coupled receptor to activate second messengers.24 Prostaglandins have a key role in maintaining physiological homeostasis as well as important roles in several diseases such as inflammatory disorders, sensory neuron-related disorders and cancer.25, 26, 27

Arachidonic acid cascade.
Although many synthetic inhibitors of prostaglandin release have been developed by targeting inhibition of COX activity, for example, nonsteroidal anti-inflammatory drugs such as indomethacin,28 new compounds with unique mechanisms of action are still needed.29
Natural products are both potential sources of probes for understanding of inflammatory mechanisms and important seed compounds for new anti-inflammatory drugs. Licochalcone A, a major phenolic constituent of the licorice species Glycyrrhiza inflate inhibits production of lipopolysaccharide (LPS)-induced PGE2 by dermal fibroblasts.30 As such, licochalcone A may be beneficial for therapeutic skin care of sensitive or irritated skin. α-Viniferin, an oligostilbene of trimeric resveratrol first isolated from Caragana chamlagu has been shown to exert an anti-inflammatory effect via inhibition of prostaglandin H2.31, 32 α-Viniferin has also been isolated from Clematis mandshurica. α-Viniferin has been shown to decrease LPS-induced production of nitric oxide and PGE2, and downregulate LPS-induced expression of proinflammatory genes, such as inducible nitric oxide synthase and COX-2, by suppressing the activity of nuclear factor kappa B via dephosphorylation of Akt/phosphoinositide 3-kinase.33 Tryptanthrin, isolated from the medicinal plant Isatis tinctoria (Brassicaceae) has also been identified as a COX-2 inhibitor.34 Triterpenoids, ursolic acid, 23-hydroxyursolic acid, 3-O-α-l-arabinopyranosyl-23-hydroxyursonic acid and 3-O-β-d-glucopyranosyl-23-hydroxyursolic acid were isolated from the stem bark of Cussonia bancoensis and evaluated for effects on LPS-induced nitric oxide and PGE2 release using the macrophage cell line RAW 264.7.35 Tectorigenin and tectoridin, isolated from the rhizomes of Korean Belamcanda chinensis (Iridaceae) for the treatment of inflammation, suppressed PGE2 production by rat peritoneal macrophages stimulated by 12-O-tetradecanoylphorbol 13-acetate (a protein kinase C (PKC) activator) or thapsigargin, a endomembrane Ca2+-ATPase inhibitor.36 Hydroxytyrosol, a major antioxidant constituent of olive oil and cardioprotective polyphenol present in Mediterranean diets, reduced matrix metallopeptidase 9 (MMP-9) and COX-2 induction in activated human monocytes via PKC-α and PKC-β1 inhibition.37
Inhibitors of prostaglandin release eliciting no effect on in vitro COX activity were sought from natural product sources. During primary screening, the inhibitory activity of samples on PGE2 release induced by bradykinin in human synovial sarcoma SW982 cells was examined. Positive samples were characterized for inhibitory activity on 6-keto-prostaglandin F1α (a stable prostacyclin (PGI2) metabolite) production, interleukin-6 release and in vitro COX-2 enzyme activity. After screening samples derived from microorganisms and plants, pronqodine A was isolated from the culture medium of Streptomyces sp. MK832-95F2.8 The structure of pronqodine A was found to comprise a benzo[d]isothiazole-4,7-dione structure using spectral analysis and total synthesis (Figures 2a and b). Pronqodine A inhibited bradykinin-induced prostaglandin release in a concentration-dependent manner, but did not affect the release of interleukin-6 from SW982 cells (Figure 2c). The IC50 values of pronqodine A for PGE2 production, 6-keto-prostaglandin F1α production and prostaglandin D2 production were 9, 17 and 7 nm, respectively.

(a) Structure of pronqodine A. (b) Synthetic scheme of pronqodine A: (i) S, NH4OH, 2-MeOCH2OH; (ii) Py-HCl, 168 °C; (iii) PIFA, EtOH; (iv) MeNH2, CeCl3, EtOH. (c) Effect of pronqodine A on 1 nm bradykinin-induced release of prostaglandin E2 (PGE2), 6-keto-prostaglandin F1α and prostaglandin D2 (PGD2) and interleukin-6 (IL-6) from SW982 cells. (d) Effect of pronqodine A and menadione as an electron acceptor in the NAD(P)H quinone dehydrogenase 1 (NQO1) activity assay. (e) Positive liquid chromatography coupled to high-resolution ESIMS spectra for pronqodine A and NQO1 reaction mixtures at m/z 195.0202–195.0242 (native form of pronqodine A) and 197.0359–197.0399 (two-electron reductive form of pronqodine A) at 0 min (upper), 2 min (middle) and 30 min (lower). Reaction mixtures contained 25 mm Tris-HCl (pH 7.6), 1 mm NADH, 3 mm FAD and 140 μg ml−1 NQO1, and 180 μm pronqodine A at 37 oC. (f) Confocal images of reactive oxygen species (ROS) production induced by pronqodine A in SW982 cells. (g) Quantitation of ROS production induced by pronqodine A and menadione in SW982 cells. (c–g) Reprinted with permission from ref. 8. Copyright (2017) American Chemical Society. A full color version of this figure is available at The Journal of Antibiotics journal online.
NAD(P)H dehydrogenase quinone 1 (NQO1) is known to catalyze the conversion of the quinone structure into its two-electron reduced form.38 Figure 2d shows the change of NADH absorbance at 340 nm in an NQO1 activity assay of pronqodine A or menadione. Menadione was converted into its two-electron reductive form menadiol (2-methyl-1,4-napthalenediol) by NQO1.39 Pronqodine A and menadione decreased the absorbance at 340 nm in a concentration-dependent manner, indicating that both compounds act as electron acceptors (Figure 2d). To identify whether pronqodine A is reduced by NQO1, a liquid chromatography coupled to high-resolution ESIMS analysis was performed on pronqodine A. The two-electron reductive form of pronqodine A converted by NQO1was identified at 2 min (Figure 2e). This peak was diminished at 30 min, while the peak of pronqodine A itself returned, suggesting that the hydroquinone form of pronqodine A was oxidized back to the quinone type. As a result of the instability of the two-electron reductive form, menadiol formed from menadione by NQO1 is known to undergo reformation to menadione with concomitant formation of reactive oxygen species (ROS) in human embryonic kidney 293 cells expressing NQO1.39 Similarly, the reductive form of pronqodine A was identified to be unstable and immediately autoxidized to its native form. Pronqodine A caused intracellular ROS production in SW982 cells (Figure 2f), and induced ROS production at a lower concentration than menadione (Figure 2g).
COX and prostaglandin synthases are well documented for their ability to convert arachidonic acid into prostaglandins such as PGE2, PGI2 and prostaglandin D2. Arachidonic acid induced the release of PGE2, which was inhibited by pronqodine A in a concentration-dependent manner (Figure 3a). Subsequently, the effect of pronqodine A on COX-1 and COX-2 expression was investigated. In SW982 cells, COX-1 expression was undetectable, while pronqodine A did not affect COX-2 expression (Figure 3b). Next, the effect of pronqodine A on COX-2 activity was examined. While treatment with pronqodine A alone did not affect COX-2 activity (Figure 3c), pronqodine A inhibited COX-2 enzyme activity in the presence of NQO1 (Figure 3d).

(a) Effect of pronqodine A on 10 μg ml−1 arachidonic acid-induced prostaglandin E2 release from SW982 cells. (b) Effect of pronqodine A on expression of cyclooxygenase 1 and 2 (COX-1 and COX-2) in SW982 cells. The concentration of bradykinin was 1 nm for 30 min. (c) Effect of pronqodine A on COX-2 enzyme activity. (d) Effect of pronqodine A with NAD(P)H quinone dehydrogenase 1 (NQO1) on COX-2 enzyme activity. (e) Generation of reactive oxygen species through redox of pronqodine A. (a–d) Reprinted with permission from ref. 8. Copyright (2017) American Chemical Society.
Taken together, NQO1 converted pronqodine A to a reduced form, which undergoes autoxidation to revert to its native form in a ROS-generating process. Thus, pronqodine A could regulate prostaglandin release by a ROS-dependent mechanism (Figure 3e).
Sacchathridin A isolated from the fermentation broth of strain Saccharothrix sp. MI559-46F5 was identified to be a PGE2 release inhibitor.9 The structure of sacchathridin A was found to be a new napthoquinone derivative with an acetylhydrazino moiety (Figure 4a). Upon examining the effect of sacchathridine A on PGE2 release from SW982 cells, inhibition was found to occur in a concentration-dependent manner with an IC50 value of 1.0 μm without affecting cell growth (Figures 4b and c). Thus, sacchathridine A could be a chemical tool for mechanistic analysis of PGE2 release.

(a) Structure of sacchathridine A. (b) Effect of sacchathridine A on prostaglandin E2 release from SW982 cells. (c) Effect of sacchathridine A on cell growth of SW982 cells.
Regulation of immune cells during inflammation
Monocytes and macrophages express VEGFR-1 (Flt-1), which has a crucial role in promoting inflammation.11, 12, 13, 14, 15, 16 Moreover, VEGFR-1 tyrosine kinase signaling modulates the proliferation of bone marrow hematopoietic cells, as well as the immunity of monocytes/macrophages and chronic inflammation.40 As such, suppression of VEGFR-1 signaling by a small molecule may significantly inhibit the underlying cause of rheumatoid arthritis.40, 41
Vegfrecine was isolated from the culture broth of Streptomyces sp. MK931-CF8 during the target-based screening of microbial metabolites for compounds with inhibitory activity against VEGFR-1 (Flt-1) tyrosine kinase. The structure of vegfrecine was determined by NMR and MS analysis combined with synthesis (Figures 5a and b).17

(a) Structure of vegfrecine. (b) Synthetic scheme of vegfrecine: (i) 2-(t-butyl-dimethylsilyloxy)aniline, NaIO4, MeOH-water, rt, 70%; (ii) 2 n NH3, CHCl3-MeOH, rt, 95%; (iii) Bu4NF, AcOH, THF, rt, 90%.
Vegfrecine exhibited potent inhibitory activity against VEGFR-1 (Flt-1) and other VEGF receptors (VEGFR-2 and -3; Table 1) and also weakly inhibited platelet-derived growth factor receptor-α and -β, fibroblast growth factor and epidermal growth factor tyrosine kinases in vitro. VEGFR-1 (Flt-1) is involved in promoting inflammation, while both VEGFR-1 (Flt-1) and VEGFR-2 (kinase insert domain receptor) are responsible for endothelial cell proliferation and blood vessel permeability. VEGFR-3 (Flt-4) participates in lymphatic vessel development. These receptor tyrosine kinases are also essential for tumor angiogenesis and lymphangiogenesis.42, 43, 44
Vegfrecine exhibited a weak inhibitory activity at concentration over 10 μm against ligand-induced phosphorylation of VEGFR-1 (Flt-1) in NIH3T3-Flt-1 cells. The inhibitory activity of vegfrecine in cells was 100-fold less than that in vitro. Notably, as vegfrecine showed poor solubility in most media, weak inhibitory activity in cells could arise from poor membrane permeability. Thus, improvement of its structural profile would be required.
Heparanase was originally characterized as a matrix degradation enzyme involved in basement membrane degradation and cancer cell invasion. Elevated expression of heparanase was linked to the malignancy and invasiveness of melanoma cells.45 Heparanase cleaves the glycosaminoglycan heparan sulfate side chain of heparan sulfate proteoglycans at the glycoside bond of glucuronic acid (Figure 6a). The involvement of heparanase in immune reactions was demonstrated by heparan sulfate-degrading activity in immune cells (neutrophils, macrophages and activated T lymphocytes) that resulted in immune cell diapedesis and accumulation within organs.46, 47, 48, 49, 50, 51, 52 A significant increase in the expression and enzymatic activity of heparanase appears in many inflammatory conditions, associated with degradation of heparan sulfate and remodeling of extracellular matrix.19, 53

(a) Heparanase cleavage site within the structure of heparan sulfate. (b) Siastatin B and heparastatin (SF4).
Heparanase inhibitors, such as heparin and heparin-mimicking compounds, exhibit anti-inflammatory effects both in vitro and in vivo. Heparin suppressed the development of experimental autoimmune encephalomyelitis (EAE).50 PI-88, also known as muparfostat, was examined for activity on eosinophil infiltration in ovalbumin-induced lung allergic inflammation.54 PI-88 reduced the number of infiltrating eosinophils. Moreover, mice with genetic elimination of heparanase exhibited reduced migration of dendritic cells55, 56, 57 and monocytes.58 Sulfated polysaccharide reduced EAE disease severity.59, 60 The inhibitory activity of chemically modified heparins, such as periodate-oxidized heparin, N-desulfated heparin and N-desulfated acetylated heparin, appear to correlate with EAE suppression.61 Effects of the heparanase enzymatic inhibitor ST1514 (glycol split, nonanticoagulant heparin) on delayed-type hypersensitivity reactions were evaluated in the ears of mice. Treatment with ST1514 resulted in decreased ear swelling and edema formation, supporting to the involvement of heparanase in delayed-type hypersensitivity.62 Enoxaparin, an analog of the syndecan-1 heparan sulfate chain, reduced the lethality of syndecan-1-deficient mice arising from dextran-induced colitis.63
Thus far, heparastatin (SF4) has been developed as a heparanase inhibitor for cancer therapy,64, 65, 66, 67 starting from siastatin B isolated as an inhibitor of sialidase (neuraminidase) from Streptomyces culture by Umezawa and Aoyagi (Figure 6b).21 Heparastatin (SF4) exhibits potent inhibitory activity (IC50=1.02±0.29 μm) against recombinant human heparanase (endo-β-glucuronidase) in human melanoma A375M cells transfected with pBK-CMV expression vectors containing heparanase cDNA.64, 68 Heparastatin (SF4) inhibited the invasion of B16BL6 and Lewis lung carcinoma (3LL) cells through reconstituted Matrigel basement membrane.63 Pulmonary colonization, after i.v. transplantation of B16BL6 cells was suppressed by heparastatin (SF4) in a dose-dependent manner.66, 67 Heparastatin (SF4) also inhibited spontaneous lung metastasis of 3LL cells.65
Considering the similarities between invasion of metastatic cancer cells and extravasation of leukocytes, application of heparanase inhibitors to suppression of inflammatory reactions is potentially significant. Recently, heparastatin (SF4) was applied to a dorsal air pouch inflammation model to demonstrate suppression of extravasation of neutrophils and monocytes by the impairment of basement membrane degradation.20 Heparastatin (SF4) suppressed local inflammation in both N-formyl-Met-Leu-Phe (fMLP) and carrageenan-induced dorsal air pouch inflammation models. Anti-inflammatory effects of heparastatin (SF4) co-administrated into the air pouch were also evaluated. Numbers of total infiltrated cells in mice treated with heparastatin (SF4) were significantly lower in fMLP-treated (67%) and, in carrageenan-treated (51%) mice compared with mice injected with proinflammatory agents alone (Figure 7a). Numbers of both neutrophils and monocytes were markedly lower in heparastatin (SF4)-injected mice. Ratios of neutrophils/monocytes were similar in the presence of heparastatin (SF4) (Figures 7b and c). However, numbers of infiltrating neutrophils were unchanged in heparastatin (SF4)-treated mice compared with control mice, suggesting heparanase was not involved in neutrophil infiltration in zymosan-induced peritonitis model (Figure 7d).

Heparastatin (SF4) suppresses infiltration of neutrophils and monocytes into inflamed air pouches in vivo. (a–c) N-formyl-Met-Leu-Phe (fMLP, 1 μm) or carrageenan (1%) in saline with or without 0.1 mm heparastatin (SF4) was injected into air pouches. After 4 h, exudates were collected and numbers of total infiltrating cells (a), neutrophils (b) and monocytes (c) were counted and plotted (n=6 per group). (d) Peritoneal exudates derived from zymosan-injected mice were collected. Numbers of neutrophils in the peritoneal fluid were counted (n=3). *P<0.05, **P<0.01. (e–g) Chemokine concentration in dorsal air pouch exudates. Concentrations of chemokines KC, MIP-2 and MCP-1 in exudates of air pouches injected with carrageenan were measured and plotted (n=6 per group). *P<0.05. (a–g) Reprinted from ref. 20. Copyright (2017) with permission from Elsevier.
Concentrations of inflammatory chemokines (KC, MIP-2 and MCP-1) in dorsal exudates collected from the air pouch model were determined. For carrageenan-mediated inflammation, there was no significant difference in tested concentrations of KC and MIP-2, while MCP-1 was significantly lower in heparastatin (SF4)-injected mice (Figures 7e–g).
Intervention by heparanase in neutrophil migration was investigated using in vitro migration and invasion tests. Migratory capacity was measured as the number of neutrophils that passed through an uncoated membrane, whereas invasion capacity was measured as the number of neutrophils that penetrated through a membrane coated with Matrigel. Addition of heparastatin (SF4) into the medium significantly lowered the number of invading neutrophils, but did not alter the number of migrating neutrophils (Figure 8). Taken together, heparastatin (SF4) significantly suppressed infiltrating monocytes and neutrophils in fMLP- or carrageenan-injected air pouches. Also, heparastatin (SF4) suppressed neutrophil invasion in vitro. Thus, this study suggested an involvement of heparanase infiltration in mouse inflammatory leukocytes, indicating a therapeutic application of heparastatin (SF4) in the suppression of inflammatory diseases.

Effect of heparastatin (SF4) on migration and invasion of neutrophils through polycarbonate membranes. Indicated concentrations of heparastatin (SF4) were added to both upper and lower chambers. Numbers of migrating (a) or invading (b) cells on the lower surface were counted after 0.5 or 24 h of incubation, respectively. Data are shown as average±s.e. N=3, *P<0.05, **P<0.01. Reprinted from ref. 20. Copyright (2017) with permission from Elsevier.
Bone repair during bone disease
Bone diseases such as inflammatory arthritis, osteoporosis and osteonecrosis are germane to the reconstitution of lost bone where processes of acute and chronic inflammation have integral roles. Dysregulated inflammation within bone causes increased bone resorption and suppressed bone formation. Crosstalk between inflammatory cells and cells related to bone healing is essential to the formation, repair and remodeling of bone.69 Cells related to bone healing consists of mesenchymal stem cells, as well as osteoblast and vascular lineages. Differentiation of mesenchymal stem cells into osteoblasts is an important process for developing bone disease therapies.
The synthetic small-molecule agonist of hedgehog signaling, purmorphamine induced ALP activity as indicated by ALP staining.70, 71, 72, 73 Natural products exhibiting osteogenic-inducing activity have recently gained much attention in addition to synthetic small molecules. Genistein, an isoflavone, has been shown to increase bone formation and decrease bone absorption.74 In addition, structurally related daidzein and formononetin were shown to induce mRNA and protein expression of BMP-2 in the human osteogenetic cell line MG63.75 Epigallocatechin-3-gallate, a green tea catechin, has a stimulatory effect on human bone marrow stem cells developing toward the osteogenic lineage as evidenced by increased ALP activity, upregulated expression of osteogenic genes and the formation of bone-like nodules.76 Honokiol, a major active ingredient of Magnolia extract, is a potent inducer of in vitro osteoblast differentiation by virtue of its capacity to suppress basal and tumor necrosis factor alpha-induced nuclear factor kappa B activation, which alleviates the suppressive action of tumor necrosis factor alpha on BMP-2 induced Smad activation.77
The mouse mesenchymal cell line C3H10T1/2 expresses mesenchymal stem cell-like pluripotency markers, thus providing a valuable model system. DPA was identified and isolated from the fermentation broth of P. verruculosum CR37010 during screening for inducers of osteoblast differentiation of pluripotent mesenchymal C3H10T1/2 cells using an early osteoblast marker (ALP activity).23 The structure of DPA was clarified to possess a tetraenoic acid and propyl side chain on the decalin ring (Figure 9a).

(a) Relative stereochemistry of decalpenic acid (DPA). (b) Induction of an early osteoblastic marker (alkaline phosphatase; ALP) activity by DPA. C3H10T1/2 cells were treated with 0.1% dimethylsulfoxide (DMSO), purmorphamine (PM) or DPA. After 4 days of treatment, ALP activities were measured as described in methods. Data are shown as mean±s.d. of three independent experiments performed in duplicate. (c) Real-time PCR analysis of osteoblastic marker genes in C3H10T1/2 cells treated with DPA. C3H10T1/2 cells were treated with 1 μm PM, 400 ng ml−1 BMP-2 (BM) or 1 μm DPA. After 8 days of treatment, total RNA was isolated from cells and mRNA expression was determined by real-time PCR analysis for Alpl, osteocalcin (Ocn) and Fabp4. Data were normalized to the level of Gapdh and are presented as fold-induction versus untreated controls. Data represent the mean±s.d. of three independent experiments. (b, c) Reproduced from ref. 23 with permission. (d) Retinoic acid response element (RARE) reporter assay. Cells were transiently transfected using a Cignal RARE reporter kit (Qiagen, Hilden, Germany). After treatment for 24 h with all-trans retinoic acid (ATRA), DPA or PM; a hedgehog signaling agonist), the dual luciferase assay was performed. RARE-luc is a reporter plasmid in which the firefly luciferase gene is under the control of tandem RAREs. NC-luc is a negative control reporter plasmid lacking the RARE element. Each error bars represents mean±s.d. obtained from three independent samples. (e) Retinoic acid receptor activation profiles of DPA. (f) Effect of retinoic acid receptor gamma knockdown on the biological activity of DPA in C3H10T1/2 cells. Twenty-four hours after Rar_gamma siRNA transfection, cells were treated with 1 μm DPA for 48 h, and mRNA levels of Alpl and Opn were analyzed by real-time reverse transcription-PCR. Each error bar represents the mean±s.d. obtained from three independent samples. (d–f) Reproduced from ref. 84 with permission Elsevier. A full color version of this figure is available at The Journal of Antibiotics journal online.
Effects of DPA on osteoblast differentiation of C3H10T1/2 cells were examined. Four-day treatment of cells with 1 μm purmorphamine induced ALP activity, as measured by ALP staining (Figure 9b). Treatment with DPA also induced ALP activity in a dose-dependent manner. The half maximal effective concentration for DPA based on ALP expression was estimated at 2 μm. Thus, DPA alone was able to induce ALP activity in C3H10T1/2 cells.
To examine whether DPA could induce the expression of other osteoblast differentiation markers, real-time PCR analysis of a panel of osteoblast marker genes was carried out. Treatment of cells with 1 μm DPA for 8 days significantly increased mRNA expression of Alpl, the gene encoding ALP but did not increase expression levels of the late osteoblast marker Ocn (Figure 9c).
C3H10T1/2 cells, like mesenchymal stem cells, can differentiate into various cell types such as adipocytes and osteoblasts, moreover, treatment of C3H10T1/2 cells with BMP-2 induced osteoblastgenesis and adipocytegenesis.78, 79 Thus, the effects of DPA on adipogenesis were examined in C3H10T1/2 cells. Treatment with BMP-2 induced adipocyte differentiation, as indicated by Fabp4 mRNA expression (Figure 9c). In contrast, treatment with 1 μm DPA or 1 μm purmorphamine did not induce this adipocyte differentiation marker. These results suggest that treatment with DPA alone did not induce adipocyte differentiation in C3H10T1/2 cells, instead inducing early osteoblastic markers through activation of signaling pathways other than Hedgehog and BMP.
Osteoblast differentiation is stimulated by retinoids.80, 81, 82, 83 Effects of DPA in C3H10T1/2 cells were compared with all-trans retinoic acid, the most important endogenous retinoid.84 A reporter assay incorporating a luciferase reporter driven by a multimerized retinoic acid response element demonstrated that DPA induced an approximately fourfold increase in luciferase activity compared with controls (Figure 9d). These data suggest that DPA acts as a retinoid to activate the retinoic acid receptor (RAR) signaling pathway in C3H10T1/2 cells.
Two families of RARs activate transcription by transducing retinoic acid signals: RARs and rexinoid receptors (RXRs).85, 86 As our results demonstrated that DPA primarily acts as a retinoid, activation of the RARs, RARα, RARβ and RARγ, as well as the RXR, RXRα, by DPA was examined. Human embryonic kidney 293T cells transiently transfected with GAL4-fusion constructs for each of the human receptors were investigated for effects of DPA, all-trans retinoic acid and 9-cis retinoic acid on transcriptional activation at the GAL4 upstream activating sequence (Figure 9e). As previously reported, 9-cis retinoic acid showed high efficiency in activating RARs and RXRα.84 All-trans retinoic acid also showed high activity against RARs and weaker activity against RXRα. In this system, DPA induced significant activation of RARγ and weak activation of RARα, whereas DPA caused no significant activation of RARβ and RXRα. These results indicated that DPA is a new retinoid that exhibits selective activity toward RARβ and RXRα.
We examined whether DPA induces early osteoblastic markers (Alpl and Opn) by activating of RARs. Early osteoblastic markers induced by DPA were inhibited in cells transfected Rarγ-targeting siRNA (Figure 9f). Thus, DPA may be used to specifically activate RARγ and modulate RARγ-related biological events.
Closing remarks
This article describes current progress in the chemical, biochemical and therapeutic potential of microbial metabolites and derivatives targeting inflammation and bone disease. Natural inhibitors affecting target enzymes or receptors associated with inflammation and bone disease have been isolated from microbial metabolites. These inhibitors and a synthetic derivative of a microbial metabolite were used for analysis of inflammatory processes. Pronqodine A inhibits release of prostaglandins (PGE2, 6-keto-prostaglandin F1α and prostaglandin D2) by interrupting the conversion of arachidonic acid to prostaglandins. NQO1 converts pronqodine A to a reduced form that undergoes autoxidation to revert to its native form, generating ROS. This ROS interrupts the process of conversion of arachidonic acid to prostaglandin H2. Sacchathridine A also inhibited PGE2 release without affecting cell growth. DPA induces early osteoblastic markers such as ALP activity and Opn mRNA in C3H10T1/2 cells via activation of RARs, especially RARγ. Thus, DPA could be used as a chemical tool for modulating biological events mediated by RARγ. Vegfrecine was discovered during the target-based screening against VEGFR-1 tyrosine kinase and exhibited selective inhibition against VEGFRs. Heparastatin (SF4) significantly suppressed infiltration of monocytes and neutrophils in an in vivo dorsal air pouch inflammation model, suggesting that cell infiltration can be pharmacologically regulated by heparastatin (SF4). It is reported that heparanase is not always involved in transmigration of immune cells, or that the involvement of heparanase is limited to cell type. Future research will focus on the involvement of cell type-dependent actions of heparanase in inflammatory processes.
Microbial metabolites have been a rich source of drug seeds for inflammation and bone disease and have thus become the special focus of research attention.
References
Aoyagi, T. [An outline of enzyme inhibitors]. Tanpakushitsu Kakusan Koso 38, 1891–1918 (1993).
Aoyagi, T., Takeuchi, T., Matsuzaki, A., Kawamura, K. & Kondo, S. Leupeptins, new protease inhibitors from Actinomycetes. J. Antibiot. (Tokyo) 22, 283–286 (1969).
Umezawa, H., Aoyagi, T., Morishima, H., Kunimoto, S. & Matsuzaki, M. Chymostatin, a new chymotrypsin inhibitor produced by actinomycetes. J. Antibiot. (Tokyo) 23, 425–427 (1970).
Aoyagi, T. et al. Poststatin, a new inhibitor of prolyl endopeptidase, produced by Streptomyces viridochromogenes MH534-30F3. I. Taxonomy, production, isolation, physico-chemical properties and biological activities. J. Antibiot. (Tokyo) 44, 949–955 (1991).
Omura, S., Ohno, H., Saheki, T., Yoshida, M. & Nakagawa, A. Elasnin a new human granulocyte elastase inhibitor produced by a strain of Streptomyces. Biochem. Biophys. Res. Commun. 83, 704–709 (1978).
Inoue, H. et al. ICM0201, a new inhibitor of osteoclastogenesis from Cunninghamella sp. F-1490. I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. (Tokyo) 56, 209–213 (2003).
Someno, T., Inoue, H., Kumagai, H., Ishizuka, M. & Takeuchi, T. ICM0201, a new inhibitor of osteoclastogenesis from Cunninghamella sp. F-1490. II. Structure determination and synthesis. J. Antibiot. (Tokyo) 56, 214–218 (2003).
Nakae, K. et al. NAD(P)H quinone oxidoreductase 1 (NQO1)-bioactivated pronqodine A regulates prostaglandin release from human synovial sarcoma cells. J. Nat. Prod. 76, 510–515 (2013).
Nakae, K. et al. Sacchathridine A, a prostaglandin release inhibitor from Saccharothrix sp. J. Nat. Prod. 76, 720–722 (2013).
Clague, R. B., Shaw, M. J. & Holt, P. J. Incidence of serum antibodies to native type I and type II collagens in patients with inflammatory arthritis. Ann. Rheum. Dis. 39, 201–206 (1980).
Adini, A., Kornaga, T., Firoozbakht, F. & Benjamin, L. E. Placental growth factor is a survival factor for tumor endothelial cells and macrophages. Cancer Res. 62, 2749–2752 (2002).
Hattori, K. et al. Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1(+) stem cells from bone-marrow microenvironment. Nat. Med. 8, 841–849 (2002).
Sawano, A. et al. Flt-1, vascular endothelial growth factor receptor 1, is a novel cell surface marker for the lineage of monocyte-macrophages in humans. Blood 97, 785–791 (2001).
Luttun, A. et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat. Med. 8, 831–840 (2002).
Lyden, D. et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 7, 1194–1201 (2001).
Hiratsuka, S., Minowa, O., Kuno, J., Noda, T. & Shibuya, M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl Acad. Sci. USA 95, 9349–9354 (1998).
Nosaka, C. et al. Vegfrecine, an inhibitor of VEGF receptor tyrosine kinases isolated from the culture broth of Streptomyces sp. J. Nat. Prod. 76, 715–719 (2013).
Vreys, V. & David, G. Mammalian heparanase: what is the message? J. Cell Mol. Med. 11, 427–452 (2007).
Meirovitz, A., Goldberg, R., Binder, A., Rubinstein, A. M., Hermano, E. & Elkin, M. Heparanase in inflammation and inflammation-associated cancer. FEBS J. 280, 2307–2319 (2013).
Sue, M. et al. An iminosugar-based heparanase inhibitor heparastatin (SF4) suppresses infiltration of neutrophils and monocytes into inflamed dorsal air pouches. Int. Immunopharmacol. 35, 15–21 (2016).
Umezawa, H., Aoyagi, T., Komiyama, T., Morishima, H. & Hamada, M. Purification and characterization of a sialidase inhibitor, siastatin, produced by Streptomyces. J. Antibiot. (Tokyo) 27, 963–969 (1974).
Rodan, G. A. & Martin, T. J. Therapeutic approaches to bone diseases. Science 289, 1508–1514 (2000).
Sakamoto, S. et al. Decalpenic acid, a novel small molecule from Penicillium verruculosum CR37010, induces early osteoblastic markers in pluripotent mesenchymal cells. J. Antibiot. (Tokyo) 63, 703–708 (2010).
Breyer, R. M., Bagdassarian, C. K., Myers, S. A. & Breyer, M. D. Prostanoid receptors: subtypes and signaling. Annu. Rev. Pharmacol. Toxicol. 41, 661–690 (2001).
Nakae, K. et al. Functional role of prostacyclin receptor in rat dorsal root ganglion neurons. Neurosci. Lett. 388, 132–137 (2005).
Cathcart, M. C., O'Byrne, K. J., Reynolds, J. V., O'Sullivan, J. & Pidgeon, G. P. COX-derived prostanoid pathways in gastrointestinal cancer development and progression: novel targets for prevention and intervention. Biochim. Biophys. Acta 1825, 49–63 (2012).
Moreira, L. & Castells, A. Cyclooxygenase as a target for colorectal cancer chemoprevention. Curr. Drug Targets 12, 1888–1894 (2011).
Ramalho, T. C., Rocha, M., da Cunha, E. F. & Freitas, M. P. The search for new COX-2 inhibitors: a review of 2002–2008 patents. Expert Opin. Ther. 9, 1193–1228 (2009).
Ng, S. C. & Chan, F. K. NSAID-induced gastrointestinal and cardiovascular injury. Curr. Opin. Gastroenterol. 26, 611–617 (2010).
Kolbe, L. et al. Anti-inflammatory efficacy of Licochalcone A: correlation of clinical potency and in vitro effects. Arch. Dermatol. Res. 298, 23–30 (2006).
Kitanaka, S. et al. (+)-Alpha-viniferin, an anti-inflammatory compound from Caragana chamlagu root. Chem. Pharm. Bull. (Tokyo). 38, 432–435 (1990).
Lee, S. H. et al. Alpha-viniferin: a prostaglandin H2 synthase inhibitor from root of Carex humilis. Planta Med. 64, 204–207 (1998).
Dilshara, M. G. et al. Anti-inflammatory mechanism of alpha-viniferin regulates lipopolysaccharide-induced release of proinflammatory mediators in BV2 microglial cells. Cell Immunol. 290, 21–29 (2014).
Danz, H. et al. Inhibitory activity of tryptanthrin on prostaglandin and leukotriene synthesis. Planta Med. 68, 875–880 (2002).
Shin, K. M. et al. In vitro anti-inflammatory activity of 23-hydroxyursolic acid isolated from Cussonia bancoensis in murine macrophage RAW 264.7 cells. Planta Med. 70, 803–807 (2004).
Kim, Y. P. et al. Inhibition by tectorigenin and tectoridin of prostaglandin E2 production and cyclooxygenase-2 induction in rat peritoneal macrophages. Biochim. Biophys. Acta 1438, 399–407 (1999).
Scoditti, E. et al. Hydroxytyrosol suppresses MMP-9 and COX-2 activity and expression in activated human monocytes via PKCalpha and PKCbeta1 inhibition. Atherosclerosis 232, 17–24 (2014).
Cadenas, E. Antioxidant and prooxidant functions of DT-diaphorase in quinone metabolism. Biochem. Pharmacol. 49, 127–140 (1995).
Nishiyama, T., Izawa, T., Usami, M., Ohnuma, T., Ogura, K. & Hiratsuka, A. Cooperation of NAD(P)H:quinone oxidoreductase 1 and UDP-glucuronosyltransferases reduces menadione cytotoxicity in HEK293 cells. Biochem. Biophys. Res. Commun. 394, 459–463 (2010).
Murakami, M. et al. Signaling of vascular endothelial growth factor receptor-1 tyrosine kinase promotes rheumatoid arthritis through activation of monocytes/macrophages. Blood 108, 1849–1856 (2006).
De Bandt, M. et al. Blockade of vascular endothelial growth factor receptor I (VEGF-RI), but not VEGF-RII, suppresses joint destruction in the K/BxN model of rheumatoid arthritis. J. Immunol. 171, 4853–4859 (2003).
Carmeliet, P. & Jain, R. K. Angiogenesis in cancer and other diseases. Nature 407, 249–257 (2000).
Saaristo, A. et al. Vascular endothelial growth factor-C and its receptor VEGFR-3 in the nasal mucosa and in nasopharyngeal tumors. Am. J. Pathol. 157, 7–14 (2000).
Karkkainen, M. J. & Petrova, T. V. Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene 19, 5598–5605 (2000).
Nakajima, M., Irimura, T., Di Ferrante, D., Di Ferrante, N. & Nicolson, G. L. Heparan sulfate degradation: relation to tumor invasive and metastatic properties of mouse B16 melanoma sublines. Science 220, 611–613 (1983).
Naparstek, Y., Cohen, I. R., Fuks, Z. & Vlodavsky, I. Activated T lymphocytes produce a matrix-degrading heparan sulphate endoglycosidase. Nature 310, 241–244 (1984).
Matzner, Y., Bar-Ner, M., Yahalom, J., Ishai-Michaeli, R., Fuks, Z. & Vlodavsky, I. Degradation of heparan sulfate in the subendothelial extracellular matrix by a readily released heparanase from human neutrophils. Possible role in invasion through basement membranes. J. Clin. Invest. 76, 1306–1313 (1985).
Fridman, R., Lider, O., Naparstek, Y., Fuks, Z., Vlodavsky, I. & Cohen, I. R. Soluble antigen induces T lymphocytes to secrete an endoglycosidase that degrades the heparan sulfate moiety of subendothelial extracellular matrix. J. Cell Physiol. 130, 85–92 (1987).
Vlodavsky, I. et al. Expression of heparanase by platelets and circulating cells of the immune system: possible involvement in diapedesis and extravasation. Invasion Metastasis 12, 112–127 (1992).
Lider, O. et al. Suppression of experimental autoimmune diseases and prolongation of allograft survival by treatment of animals with low doses of heparins. J. Clin. Invest. 83, 752–756 (1989).
Lider, O. et al. Inhibition of T lymphocyte heparanase by heparin prevents T cell migration and T cell-mediated immunity. Eur. J. Immunol. 20, 493–499 (1990).
Yahalom, J., Fibach, E., Bar-Tana, R., Fuks, Z. & Vlodavsky, I. Differentiating human leukemia cells express heparanase that degrades heparan sulfate in subendothelial extracellular matrix. Leuk. Res. 12, 711–717 (1988).
Li, J. P. & Vlodavsky, I. Heparin, heparan sulfate and heparanase in inflammatory reactions. Thromb. Haemost. 102, 823–828 (2009).
Morris, A. et al. The role of heparanase in pulmonary cell recruitment in response to an allergic but not non-allergic stimulus. PLoS ONE 10, e0127032 (2015).
Benhamron, S. et al. Dissociation between mature phenotype and impaired transmigration in dendritic cells from heparanase-deficient mice. PLoS ONE 7, e35602 (2012).
Poon, I. K. et al. Mice deficient in heparanase exhibit impaired dendritic cell migration and reduced airway inflammation. Eur. J. Immunol. 44, 1016–1030 (2014).
Sasaki, N. et al. Cell surface localization of heparanase on macrophages regulates degradation of extracellular matrix heparan sulfate. J. Immunol. 172, 3830–3835 (2004).
Stoler-Barak, L. et al. Heparanase of murine effector lymphocytes and neutrophils is not required for their diapedesis into sites of inflammation. FASEB J. 29, 2010–2021 (2015).
Willenborg, D. O. & Parish, C. R. Inhibition of allergic encephalomyelitis in rats by treatment with sulfated polysaccharides. J. Immunol. 140, 3401–3405 (1988).
Willenborg, D. O. & Parish, C. R. Inhibition of passive allergic encephalomyelitis by sulfated polysaccharides. Ann. N. Y. Acad. Sci. 540, 543–545 (1988).
Parish, C. R., Hindmarsh, E. J., Bartlett, M. R., Staykova, M. A., Cowden, W. B. & Willenborg, D. O. Treatment of central nervous system inflammation with inhibitors of basement membrane degradation. Immunol. Cell Biol. 76, 104–113 (1998).
Edovitsky, E., Lerner, I., Zcharia, E., Peretz, T., Vlodavsky, I. & Elkin, M. Role of endothelial heparanase in delayed-type hypersensitivity. Blood 107, 3609–3616 (2006).
Floer, M. et al. Enoxaparin improves the course of dextran sodium sulfate-induced colitis in syndecan-1-deficient mice. Am. J. Pathol. 176, 146–157 (2010).
Nishimura, Y. et al. Flexible synthesis and biological activity of uronic acid-type gem-diamine 1-N-iminosugars: a new family of glycosidase inhibitors. J. Org. Chem. 65, 2–11 (2000).
Nishimura, Y. et al. Effect on spontaneous metastasis of mouse Lewis lung carcinoma by a trifluoroacetamide analogue of siastatin B. J. Antibiot. (Tokyo) 47, 840–842 (1994).
Nishimura, Y. et al. Totally synthetic analogues of siastatin B. III. Trifluoroacetamide analogues having inhibitory activity for tumor metastasis. J. Antibiot. (Tokyo) 47, 101–107 (1994).
Satoh, T. et al. Synthesis and antimetastatic activity of 6-trichloroacetamido and 6-guanidino analogues of siastatin B. J. Antibiot. (Tokyo) 49, 321–325 (1996).
Toyoshima, M. & Nakajima, M. Human heparanase. Purification, characterization, cloning, and expression. J. Biol. Chem. 274, 24153–24160 (1999).
Loi, F., Cordova, L. A., Pajarinen, J., Lin, T. H., Yao, Z. & Goodman, S. B. Inflammation, fracture and bone repair. Bone 86, 119–130 (2016).
Wu, X., Ding, S., Ding, Q., Gray, N. S. & Schultz, P. G. A small molecule with osteogenesis-inducing activity in multipotent mesenchymal progenitor cells. J. Am. Chem. Soc. 124, 14520–14521 (2002).
Wu, X., Walker, J., Zhang, J., Ding, S. & Schultz, P. G. Purmorphamine induces osteogenesis by activation of the hedgehog signaling pathway. Chem. Biol. 11, 1229–1238 (2004).
Beloti, M. M., Bellesini, L. S. & Rosa, A. L. Purmorphamine enhances osteogenic activity of human osteoblasts derived from bone marrow mesenchymal cells. Cell Biol. Int. 29, 537–541 (2005).
Sinha, S. & Chen, J. K. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat. Chem. Biol. 2, 29–30 (2006).
Zhou, S. et al. Estrogens activate bone morphogenetic protein-2 gene transcription in mouse mesenchymal stem cells. Mol. Endocrinol. 17, 56–66 (2003).
Li, X. et al. Identification of upregulators of BMP2 expression via high-throughput screening of a synthetic and natural compound library. J. Biomol. Screen. 14, 1251–1256 (2009).
Jin, P., Wu, H., Xu, G., Zheng, L. & Zhao, J. Epigallocatechin-3-gallate (EGCG) as a pro-osteogenic agent to enhance osteogenic differentiation of mesenchymal stem cells from human bone marrow: an in vitro study. Cell Tissue Res. 356, 381–390 (2014).
Yamaguchi, M., Arbiser, J. L. & Weitzmann, M. N. Honokiol stimulates osteoblastogenesis by suppressing NF-kappaB activation. Int. J. Mol. Med. 28, 1049–1053 (2011).
Yamaguchi, A. Regulation of differentiation pathway of skeletal mesenchymal cells in cell lines by transforming growth factor-beta superfamily. Semin. Cell Biol. 6, 165–173 (1995).
Hata, K. et al. Differential roles of Smad1 and p38 kinase in regulation of peroxisome proliferator-activating receptor gamma during bone morphogenetic protein 2-induced adipogenesis. Mol. Biol. Cell 14, 545–555 (2003).
Makhijani, N. S., Bischoff, D. S. & Yamaguchi, D. T. Regulation of proliferation and migration in retinoic acid treated C3H10T1/2 cells by TGF-beta isoforms. J. Cell Physiol. 202, 304–313 (2005).
Wiper-Bergeron, N., St-Louis, C. & Lee, J. M. CCAAT/Enhancer binding protein beta abrogates retinoic acid-induced osteoblast differentiation via repression of Runx2 transcription. Mol. Endocrinol. 21, 2124–2135 (2007).
Wang, A., Ding, X., Sheng, S. & Yao, Z. Retinoic acid inhibits osteogenic differentiation of rat bone marrow stromal cells. Biochem. Biophys. Res. Commun. 375, 435–439 (2008).
Zhang, W. et al. Retinoic acids potentiate BMP9-induced osteogenic differentiation of mesenchymal progenitor cells. PLoS ONE 5, e11917 (2010).
Sakamoto, S., Kojima, F., Momose, I., Kawada, M., Adachi, H. & Nishimura, Y. Decalpenic acid induces early osteoblastic markers in pluripotent mesenchymal cells via activation of retinoic acid receptor gamma. Biochem. Biophys. Res. Commun. 422, 751–757 (2012).
Bastien, J. & Rochette-Egly, C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 328, 1–16 (2004).
Germain, P. et al. International Union of Pharmacology. LXIII. Retinoid X receptors. Pharmacol. Rev. 58, 760–772 (2006).
Acknowledgements
We are deeply indebted to Professor M Shibasaki and Dr M Kawada for valuable discussions. We thank all past and present co-workers whose names were cited in references for their valued and enthusiastic contributions. We are grateful to Drs I Momose and N Hosokawa, and Ms F Kojima for biological studies; Dr R Sawa and Ms Y Kubota for structure determination; and Drs M Igarashi and M Hatano for microbial resources.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Dedication: We are pleased to dedicate this review article to late Professor Hamao Umezawa for his 103 birth years, 31 years after death and a devoted special issue in 2018. He was a pioneer in the field of antibiotic chemotherapy. He was awarded a number of prizes including Japanese Academy award and Paul Ehrlich prize, and many honors including the Japanese Order of Culture and Légion d’honneur. We are deeply grateful for his achievements still giving a number of study guidance to us.
Rights and permissions
About this article

Cite this article
Adachi, H., Nakae, K., Sakamoto, S. et al. Microbial metabolites and derivatives targeted at inflammation and bone diseases therapy: chemistry, biological activity and pharmacology. J Antibiot 71, 60–71 (2018). https://doi.org/10.1038/ja.2017.138
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2017.138
This article is cited by
-
Structure-activity relationships of natural quinone vegfrecine analogs with potent activity against VEGFR-1 and -2 tyrosine kinases
The Journal of Antibiotics (2021)
