
Abstract
The enzymes responsible for biotin biosynthesis in mycobacteria have been considered as potential drug targets owing to the important role in infection and cell survival that the biotin synthetic pathway plays in Mycobacterium tuberculosis. Among the enzymes that comprise mycobacterium biotin biosynthesis systems, 7,8-diaminopelargonic acid synthase (DAPAS) plays an essential role during the stationary phase in bacterial growth. In this study, compounds that inhibit mycobacterial DAPAS were screened in the virtual chemical library using an in silico structure-based drug screening (SBDS) technique, and the antimycobacterial activity of the selected compounds was validated experimentally. The DOCK–GOLD programs utilized by in silico SBDS facilitated the identification of a compound, referred to as KMD6, with potent inhibitory effects on the growth of model mycobacteria (M. smegmatis). The subsequent compound search, which was based on the structural features of KMD6, resulted in identification of three additional active compounds, designated as KMDs3, KMDs9 and KMDs10. The inhibitory effect of these compounds was comparable to that of isoniazid, which is a first-line antituberculosis drug. The high antimycobacterial activity of KMD6, KMDs9 and KMDs10 was maintained on the experiment with M. tuberculosis. Of the active compounds identified, KMDs9 would be a promising pharmacophore, owing to its long-term antimycobacterial effect and lack of cytotoxicity.
Similar content being viewed by others


Introduction
Tuberculosis (TB), which is caused by Mycobacterium tuberculosis (M. tuberculosis) infection, has been regarded as the most severe respiratory infection worldwide.1, 2, 3, 4 An increase in TB patients has been attributed to insufficient supply or low quality of anti-TB medicines as well as the emergence of drug-resistant strains, including multi-drug-resistant TB (MDR-TB) and extensively-drug-resistant TB (XDR-TB). Co-infection with HIV has also complicated tuberculosis treatment.1, 5, 6 Therefore, there is an urgent need for additional antitubercular agents with new mechanisms of action.
The enzymes responsible for biotin (vitamin H or vitamin B7) biosynthesis have been considered as potential drug targets because the biotin synthetic pathway plays an important role in the infection process and is essential for survival of M. tuberculosis.7, 8 Moreover, mammalian hosts do not have enzymes for the biosynthesis of biotin cofactor.9 M. tuberculosis synthesizes biotin from pimeloyl-CoA via four sequential enzymatic reactions.10 The second reaction, that is, the synthesis of 7,8-diaminopelargonic acid (DAPA) from 7-keto-8-aminopelargonic acid and S-adenosyl-L-methionine, is catalyzed by 7,8-diaminopelargonic acid synthase (DAPAS).8, 9 Keer et al.11 showed that DAPAS in mycobacterium is essential for de novo biotin biosynthesis during the stationary phase and that mycobacterium in this phase are unable to uptake exogenous biotin. Thus, a compound capable of binding to a mycobacterial DAPAS active site and/or a substrate-binding groove is expected to be a novel agent with original antimycobacterial action.
In recent decades, developments in computational chemistry and three-dimensional protein structure determination by X-ray crystallography or NMR have accelerated in silico screening of chemical compound libraries; this is referred to as in silico structure-based drug screening (SBDS). The SBDS technique facilitates a more rapid identification of hit compounds than ordinal screening in biological assays. To date, we have screened inhibitors for mycobacterial enzymes (for example, enoyl-acyl carrier protein reductase12, 13, 14 and thymidine monophosphate kinase15) using the in silico SBDS strategy, and the antimycobacterial action of the hit compounds has been experimentally validated. In this study, in silico SBDS was applied to search a huge virtual compound library for the most optimal compounds that could interact with the DAPAS active site and the antimycobacterial activity of the selected compounds was examined.
Materials and methods
Preparation of compound library and protein structural data
The virtual chemical library comprising 461 397 compounds and supplied by ChemBridge (San Diego, CA, USA) was prepared as previously described.12 The two-dimensional structure of each compound was converted into its three-dimensional structure using the stereochemistry module in the molecular operating environment (MOE) (Chemical Computing Group, Montreal, Canada). The protonation states were adjusted at pH 7.0 and 10-multiple conformations were generated using the MOE protonate 3D and conformation search modules. In order to screen a substrate competitive inhibitor using the SBDS strategy, utilization of competitive inhibitor bound or holo-form protein structures is preferable rather than that of apo-form. Then, the PDB structural data of the H315R-mutated (random mutation) M. tuberculosis DAPAS complex with the unreactive SAM analog sinefungin (PDB-id 3LV2)9 was employed for the present SBDS, and hydrogen atoms were added using the protonate 3D module at pH 7.0 in the MOE software after removal of sinefungin from the structure. Adjustment of the partial charges and the successive energy minimization of the protein structure were performed using the MOE partial charged and the energy minimize modules, respectively.
In silico structure-based drug screening (SBDS)
In silico drug screening was attempted with a combination of the UCSF DOCK program version 6.3,16 and GOLD program,17, 18 as previously described.12, 13, 15, 19 High-throughput DOCK screening was performed to estimate potential binding affinities using the scoring function: Eint=Evdw+Eelec, where Eint is interaction energy, Evdw is van der Waals contact energy, and Eelec is electrostatic energy. The top 1000 compounds with a DOCK score of less than −51 kcal mol−1 were selected from a small compound library (containing 461 397 compounds), and these compounds were subsequently screened using the GOLD software. For the top 100 compounds with highest GOLD scores, 10 conformations of each compound were generated by the LowModeMD20 and were rescreened using the GOLD program. The structurally related compounds were searched within the Hit2Lead database (ChemBridge) that comprises over 700 000 compounds, as previously described.13, 14, 19 Ten-conformations of the 29 compounds with a high Tanimoto coefficient (>0.85) were generated, and then, docking simulation was performed in the GOLD program.
Candidate compounds
Candidate compounds that were identified by SBDS in the present study were purchased from ChemBridge. The candidate compounds and their GOLD scores are summarized in Tables 1 and 2. The NMR and/or LC-MS data related to the compounds are available on the manufacture’s website at http://www.chembridge.com. Hereafter, the series of the screened compounds are referred to as KMD1–KMD7 and KMDs1–KMDs10.
Antimicrobial assay
M. smegmatis (IAM 12065 strain, RIKEN BioResource Center, Saitama, Japan) or M. tuberculosis (MC2 7000 strain) cultures were grown at 37 °C for 24 h in 3.7% brain heart infusion broth (Sigma, St. Louise, MO, USA). Cultures were then diluted eight-fold with broth that contained the candidate compounds in a 96-well flat-bottom clear plates (CORNING, Corning, NY, USA). Each well was inoculated with 200 μl of culture. Isoniazid (LKT laboratories, St. Paul, MN, USA) and 0.3% DMSO were used as positive and negative controls, respectively. The plates were incubated at 37 °C for 24 h, and then, the cell cultures were subjected to growth inhibition assays. Inhibition of bacterial growth was determined by measuring OD595 using Bio-Rad Model 680 micro-plate reader (Bio-Rad, Hercules, CA, USA).
Cytotoxic assay
SH-SY5Y human neuroblastoma cell lines were maintained in high-glucose Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U ml−1 of penicillin, and 100 μg ml−1 of streptomycin and cultured at 37 °C in a humidified atmosphere containing 5% CO2. SH-SY5Y cells (1.5 × 104 cells per well) were seeded into 96-well plates 24 h before serum starvation, with 0.25% FBS containing DMEM. After 48 h of serum starvation, the candidate compounds (50 μM) were added. DMSO (0.3%) was used as the negative control. The cultures were incubated for additional 48 h, and then, the cytotoxicity of the compounds was analyzed with a Cell Counting Kit-8 (Dojin, Kumamoto, Japan), as previously described.12
Results
Identification of the candidate compounds by SBDS
In this study, in silico SBDS of a 461 397 compound library was performed to identify DAPAS-targeted novel antiinfectives for M. tuberculosis. The left panel of Figure 1 shows a schematic representation of the three-step screening process by the DOCK (first screening) and GOLD programs (second screening: single conformation, third screening: multiple conformations). The primary DOCK screening identified the top 1000 ranked chemical compounds with a DOCK score of less than −51 kcal mol−1. The subsequent GOLD screenings resulted in the identification of seven compounds, designated as KMD1–KMD7, with high GOLD scores (average 84.5) (Table 1).

Schematic representation of in silico SBDS for identification of DAPAS-targeted novel antiinfectives for M. tuberculosis. Left panel: screening of candidate compounds in ChemBridge compound library (461 397) through DOCK–GOLD tandem screening as described in the 'Materials and methods' section. Right panel: screening of compounds that are structurally related to KMD6 in the Hit2Lead database. Ten-conformations of the 29 compounds with a high Tanimoto coefficient (>0.85) were generated, and then, docking simulation was performed in the GOLD program.
Growth inhibition of M. smegmatis by the candidate compounds: KMD1–KMD7
The inhibitory effect of each of the seven candidate compounds (KMD1–KMD7) on mycobacterial growth was measured. In this experiment, non-pathogenic M. smegmatis (biosafety level 1) was utilized as a model mycobacteria because the primary structures of DAPAS in M. smegmatis and M. tuberculosis are likely sufficiently similar (84%). Figure 2a shows the effects of the candidate compounds on the growth of M. smegmatis. KMD1 and KMD2 did not inhibit the bacterial growth, whereas the other compounds showed significant inhibitory effects. In particular, KMD6 strongly inhibited bacterial growth, and its effect was comparable to that of isoniazid.

Antimicrobial activity of the candidate compounds of SBDS on the growth of M. smegmatis. The inhibition of bacterial growth was monitored by OD595 at 0 and 24 h after the treatment using compounds, as described in 'materials and methods' section. The concentration of the tested compounds was 100 μM. All experiments were performed in quadruplicate and the values obtained in the absence (−) and presence (+) of tested compounds were compared using the Bonferroni’s test for significance: n.s., not significant; *P<0.05; **P<0.01; ***P<0.001. (a) Antimicrobial activities of KMD1–KMD7. Isoniazid was used as the positive control. (b) Antimicrobial activities of KMDs1–KMDs10. A full color version of this figure is available at The Journal of Antibiotics journal online.
Structure–activity relationship study of KMD6
Among the seven candidate compounds tested, KMD6 showed remarkable inhibitory effects on the growth of the model mycobacterium. To further study the structure–activity relationships of KMD6, structurally related compounds were screened within the Hit2Lead database that comprises over 700 000 compounds. The right panel of Figure 1 illustrates the screening process for compounds that possessed structural features of KMD6. From the search, ten structurally related compounds (KMDs1–KMDs10) were identified (Table 2). As described above, the inhibitory effects of KMDs1–KMDs10 (100 μM) on the growth of M. smegmatis were examined (Figure 2b). Among the tested compounds, KMDs3 had the strongest inhibitory effect on growth. Although compared with KMDs3, KMDs9 and KMDs10 induced only modest growth inhibition (~40%), the inhibitory effects of these compounds were comparable to that of isoniazid. Thus, KMD6, KMDs3, KMDs9 and KMDs10 are regarded as active compounds and expected to be novel antiinfectives for mycobacteria.
Antimycobacterial properties of the active compounds
The chemical structures of KMD6, KMDs3, KMDs9 and KMDs10 are illustrated in Figure 3a, and the dose dependence of each compound’s inhibitory effect is shown in Figure 3b. The concentration of compounds that exert a 50% inhibition on M. smegmatis growth (IC50 values) was: KMD6, 18.4 μM; KMDs3, 9.3 μM; KMDs9, 15.9 μM; and KMDs10, 14.9 μM. IC50 value of isoniazid (positive control) was 5.4 μm (Supplementary Figure S1).

Antimicrobial activities of the active compounds on the growth of M. smegmatis. (a) Chemical structures of the active compounds (KMD6, KMDs3, KMDs9 and KMDs10). (b) Dose dependence of the active compounds on the growth of M. smegmatis, where the upper left panel indicates KMD6; upper right, KMDs3; lower left, KMDs9 and lower right, KMDs10. The inhibition of bacterial growth was monitored by OD595 at 24 h after treatment with the compounds. All experiments were performed in quadruplicate. (c) Time course of the effect of active compounds on the growth of M. smegmatis. Inhibition of bacterial growth was monitored by OD595 at 24 h after treatment with the compounds. All experiments were performed in quadruplicate. The concentrations of the tested compounds were 100 μM. Isoniazid was used as the positive control. A full color version of this figure is available at The Journal of Antibiotics journal online.
Simultaneously, bacterial growth inhibition was monitored over time to validate the stability and persistence of the compounds (Figure 3c). Over the course of 72 h, KMDs3, KMDs9 and KMDs10 continually inhibited bacterial growth at the same level as isoniazid. However, the growth inhibitory effect of KMD6 was not maintained throughout the incubation.
Growth inhibition of M. tuberculosis by the active compounds
The inhibitory effect of KMD6, KMDs3, KMDs9 and KMDs10 on the growth of M. tuberculosis (MC2 7000 strain) was examined to confirm whether the compounds can inhibit the growth of pathogenic mycobacteria. Figure 4 shows the dose dependence of each compound on growth inhibition of M. tuberculosis. The IC50 values of KMD6, KMDs3, KMDs9 and KMDs10 were 2.5 μM, 48.8 μM, 3.8 μM and 8.8 μM, respectively. The IC50 value of KMDs3 was lower when tested with M. smegmatis than that when tested with M. tuberculosis, although the antimicrobial activity of the compound was retained when tested with M. tuberculosis.

Dose dependence of the active compounds (KMD6, KMDs3, KMDs9 and KMDs10) on the growth of M. tuberculosis (MC2 7000 strain), where the (a) KMD6; (b) KMDs3; (c) KMDs9; and (d) KMDs10. The inhibition of bacterial growth was monitored by OD595 at 24 h after treatment with the compounds. All experiments were performed in quadruplicate.
Cytotoxicity of the active compounds
The cytotoxicity of KMD6, KMDs3, KMDs9 and KMDs10 was measured on the SH-SY5Y cell line to evaluate whether the compounds have a damaging effect on mammalian cells (Figure 5). Only KMDs9 (50 μM) showed modest and non-significant cytotoxicity, whereas the other compounds KMD6, KMDs3 and KMDs10 exhibited stronger cytotoxic effect. Nevertheless, oral rat LD50 values of the compounds, estimated by the Toxicity Estimation Software Tool (United States Environmental Protection Agency), were >1000 mg kg−1 (estimated LD50 values for KMD6, KMDs3, KMDs9 and KMDs10 were 1,227, 1,884, 1,675 and 1,983 mg kg−1, respectively). KMD6, KMDs3 and KMDs10 exhibited cytotoxicity, though low oral toxicity of the compounds was expected.

Cytotoxicity of the active compounds (KMD6, KMDs3, KMDs9 and KMDs10) against the SH-SY5Y human neuroblastoma cell line. The concentration of the compounds was 50 μM. In total, 0.3% DMSO was used as positive control. All experiments were performed in quadruplicate, and cell survival rates were compared using the Dunnett’s test for significance: n.s., P>0.05; ***P<0.001.
Discussion
Among the enzymes essential in a biological process of M. tuberculosis, DAPAS has been defined as one of the promising target because disruption of biotin biosynthesis pathway results in cell death rather than growth arrest. In the present study, a 416 397-compound library was screened for small compounds capable of inhibiting mycobacterium DAPAS using DOCK–GOLD combined with the three-step in silico SBDS. The screening resulted in the identification of the seven candidate compounds (KMD1–7) and the all the compounds, with the exceptions of KMD1 and KMD2, exhibited growth inhibition of model mycobacteria. Additional screening based on structural similarity to the compound that showed an excellent inhibitory effect on mycobacterial growth (KMD6) identified three additional compounds (KMDs3, KMDs9 and KMDs10) with potent antimycobacterial activity. Overall, we identified four promising antiinfectives for mycobacteria out of the 17 compounds tested (24%). In general, thousands of compounds are tested in a random high-throughput screening, and the hit rate of this method is 1–3%; thus, the in silico SBDS performed in this study could be a powerful cost- and labor-saving tool in drug screening.
In our previous reports, we utilized model mycobacteria: M. smegmatis and M. vanbaalenni, to assess antimycobacterial activity of candidate compounds.12, 13, 14, 15 In the present study, we checked antimycobacterial activity of the four active compounds using M. tuberculosis. As expected, the antymycobacterial activity of active compounds was confirmed through the experiments with M. tuberculosis. Nevertheless, IC50 values of KMDs3 were different between the experiments with M. smegmatis and M. tuberculosis. Although this clear discrepancy between M. smegmatis and M. tuberculosis should be interesting, we did not go further into the detail in this study. We suppose the difference could be ascribed to the subtle structural difference of DAPAS and/or difference of membrane permeability of the compounds.
Although the four active compounds were expected to interact with the active site cavity and/or the substrate-binding groove of mycobacterium DAPAS, details of the inhibitory mechanism (for example, a target-enzyme specificity, biophysical binding properties, and effect on the enzyme kinetics and inhibiting mechanism) have not been validated in this study. This is because we endeavored to acquire a novel antimycobacterial pharmacophore through the in silico SBDS in this study and that elucidation of an inhibitory mechanism remains a goal for future work. Because prevention of MDR/XDR-TB is a top priority for global TB control, we believe that an accumulation of knowledge of the pharmacophore that addresses mycobacterium growth inhibition would help develop novel antiinfectives for M. tuberculosis.
Acidomycin (MIC=0.0625–0.125 μg ml−1),9 representative DAPAS inhibitor, has chemical structure of oxothiazolidine fused caproic acid. Interestingly, a characteristic dioxothiazolidine groups are found in the four active compounds as a consensus component. In addition, sulfonic acid ester groups, distanced by 3–5 carbons from the dioxothiazolidine groups, are also found in the all active compounds. The sulfonic acid ester groups might have similar functions of anionic carboxyl group in acydomycin. These structural similarities to acidomycin might provide insight into the antimycobacterial mechanism of the active compounds.
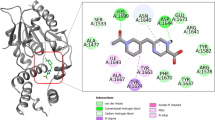
The active site of M. tuberculosis DAPAS is 24 Å deep and the substrate-binding loop comprises Pro24–Ser34, Met87–His97, Arg156–Asp160, Gln224–Gly228, Ala307–Asn322 and Arg400–Arg403.9, 21 The binding interactions between M. tuberculosis DAPAS and the active compounds KMD6, KMDs3, KMDs9 and KMDs10 were predicted with the MOE protein–ligand interaction fingerprint and ligand interaction programs (Figure 6). According to the prediction, the four active compounds interact with Arg403 in the substrate-binding loop (KMD6, KMDs3 and KMDs9: arene–H interaction, KMDs10: hydrogen bond), suggesting that the interaction of Arg403 in M. tuberculous DAPAS is essential for disrupting DAPAS activity. Eliot et al.22 concluded that Arg391 of Escherichia coli DAPAS is important for recognition of DAPA. Because Arg391 of E. coli DAPAS corresponds to Arg400 of M. tuberculosis DAPAS, the binding of the compounds to (Arg400 proximal) Arg403 in M. tuberculosis DAPAS may interfere with DAPA recognition.

Predicted binding of KMD6, KMDs3, KMDs9 and KMDs10 to active site in M. tuberculosis DAPAS by MOE protein–ligand interaction fingerprint and ligand interaction programs. A full color version of this figure is available at The Journal of Antibiotics journal online.
To date, only a limited effort to develop a M. tuberculosis DAPAS inhibitor has been reported, and DAPAS-targeted antiinfectives have not been approved. For instance, amiclenomycin, a naturally occurring inhibitor of DAPAS, and its derivative showed good antimycobacterial activity (MIC=3.1 μg ml−1);9 however, an in vivo administration experiment using amiclenomycin failed owing to its low chemical stability.23, 24 KMDs9 would be one of the most promising pharmacophores owing to its high and persistent antimycobacterial activity, and low cytotoxicity. These properties would be advantageous for long-term administration to treat MDR-TB and XDR-TB.
References
Dye, C. & Williams, B. G. The population dynamics and control of tuberculosis. Science 328, 856–861 (2010).
Lönnroth, K. et al. Tuberculosis control and elimination 2010-50: cure, care, and social development. Lancet 375, 1814–1829 (2010).
Das, P. & Horton, R. Tuberculosis–time to accelerate progress. Lancet 375, 1755–1757 (2010).
Das, P. & Horton, R. Tuberculosis–getting to zero. Lancet 386, 2231–2232 (2015).
Gandhi, N. R. et al. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet 375, 1830–1843 (2010).
Tang, J., Yam, W. C. & Chen, Z. Mycobacterium tuberculosis infection and vaccine development. Tuberculosis 98, 30–41 (2016).
Sassetti, C. M. & Rubin, E. J. Genetic requirements for mycobacterial survival during infection. Proc. Natl Acad. Sci. USA 100, 12989–12994 (2003).
Woong Park, S. et al. Evaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expression. PLoS Pathog. 7, e1002264 (2011).
Dey, S., Lane, J. M., Lee, R. E., Rubin, E. J. & Sacchettini, J. C. Structural characterization of the Mycobacterium tuberculosis biotin biosynthesis enzymes 7,8-diaminopelargonic acid synthase and dethiobiotin synthetase. Biochemistry 49, 6746–6760 (2010).
Cole, S. T. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 (1998).
Keer, J., Smeulders, M. J., Gray, K. M. & Williams, H. D. Mutants of Mycobacterium smegmatis impaired in stationary-phase survival. Microbiology 146, 2209–2217 (2000).
Izumizono, Y., Arevalo, S., Koseki, Y., Kuroki, M. & Aoki, S. Identification of novel potential antibiotics for tuberculosis by in silico structure-based drug screening. Eur. J. Med. Chem. 46, 1849–1856 (2011).
Kinjo, T. et al. Identification of compounds with potential antibacterial activity against Mycobacterium through structure-based drug screening. J. Chem. Inf. Model. 53, 1200–1212 (2013).
Kanetaka, H. et al. Discovery of InhA inhibitors with anti-mycobacterial activity through a matched molecular pair approach. Eur. J. Med. Chem. 94, 378–385 (2015).
Koseki, Y., Kinjo, T., Kobayashi, M. & Aoki, S. Identification of novel antimycobacterial chemical agents through the in silico multi-conformational structure-based drug screening of a large-scale chemical library. Eur. J. Med. Chem. 60, 333–339 (2013).
Lang, P. T. et al. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA 15, 1219–1230 (2009).
Jones, G., Willett, P. & Glen, R. C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 245, 43–53 (1995).
Jones, G., Willett, P., Glen, R. C., Leach, A. R. & Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267, 727–748 (1997).
Kobayashi, M. et al. Identification of novel potential antibiotics against Staphylococcus using structure-based drug screening targeting dihydrofolate reductase. J. Chem. Inf. Model. 54, 1242–1253 (2014).
Labute, P. LowModeMD–implicit low-mode velocity filtering applied to conformational search of macrocycles and protein loops. J. Chem. Inf. Model. 50, 792–800 (2010).
Mann, S., Marquet, A. & Ploux, O. Inhibition of 7,8-diaminopelargonic acid aminotransferase by amiclenomycin and analogues. Biochem. Soc. Trans. 33, 802–805 (2005).
Eliot, A. C., Sandmark, J., Schneider, G. & Kirsch, J. F. The dual-specific active site of 7,8-diaminopelargonic acid synthase and the effect of the R391A mutation. Biochemistry 41, 12582–12589 (2002).
Hotta, K., Kitahara, T. & Okami, Y. Studies of the mode of action of amiclenomycin. J. Antibiot. 28, 222–228 (1975).
Sandmark, J., Mann, S., Marquet, A. & Schneider, G. Structural basis for the inhibition of the biosynthesis of biotin by the antibiotic amiclenomycin. J. Biol. Chem. 277, 43352–43358 (2002).
Acknowledgements
This work was supported in part by a grant from Takeda Science Foundation to JT and a Grants-in-Aid for Young Scientists (B) (16K21226) and a Grant-in-Aid for Scientific Research (C) (26460145) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Taira, J., Morita, K., Kawashima, S. et al. Identification of a novel class of small compounds with anti-tuberculosis activity by in silico structure-based drug screening. J Antibiot 70, 1057–1064 (2017). https://doi.org/10.1038/ja.2017.106
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2017.106
This article is cited by
-
Identification of novel inhibitors for mycobacterial polyketide synthase 13 via in silico drug screening assisted by the parallel compound screening with genetic algorithm-based programs
The Journal of Antibiotics (2022)
-
Improvement of the novel inhibitor for Mycobacterium enoyl-acyl carrier protein reductase (InhA): a structure–activity relationship study of KES4 assisted by in silico structure-based drug screening
The Journal of Antibiotics (2020)
-
Mimicking the human environment in mice reveals that inhibiting biotin biosynthesis is effective against antibiotic-resistant pathogens
Nature Microbiology (2019)

{kind=link}