
Abstract
There is growing global recognition that the continued emergence of multidrug-resistant bacteria poses a serious threat to human health. Action plans released by the World Health Organization and governments of the UK and USA in particular recognize that discovering new antibiotics, particularly those with new modes of action, is one essential element required to avert future catastrophic pandemics. This review lists the 30 antibiotics and two β-lactamase/β-lactam combinations first launched since 2000, and analyzes in depth seven new antibiotics and two new β-lactam/β-lactamase inhibitor combinations launched since 2013. The development status, mode of action, spectra of activity and genesis (natural product, natural product-derived, synthetic or protein/mammalian peptide) of the 37 compounds and six β-lactamase/β-lactam combinations being evaluated in clinical trials between 2013 and 2015 are discussed. Compounds discontinued from clinical development since 2013 and new antibacterial pharmacophores are also reviewed.
Similar content being viewed by others
Introduction
Antibiotics cure disease and save lives. No other class of drugs has so cheaply and effectively prevented death from life-threatening illnesses for over 70 years. Unfortunately, antibiotics are dramatically undervalued by society, receiving a fraction of the yearly revenue per patient generated by next-generation anticancer drugs. It is well documented that antibiotics are becoming an ‘endangered species’, with older antibiotics rendered impotent by the rise of multidrug-resistant bacteria, and new antibiotics a scarce commodity due to the exit of most major pharmaceutical companies from antibiotic research.1 Fortunately, since our previous reviews,2, 3 there has been a significant shift in government and public recognition of the potential threat posed by the loss of effective therapies to treat bacterial infections, with well-publicized reports from the World Health Organization,4, 5 and national governments of countries including the UK,6, 7, 8 the USA,9, 10, 11, 12, 13 Canada14, 15 and Australia,16 among others. Government agencies are incorporating the threat of antibiotic resistance into natural disaster planning scenarios.17
A range of reviews and discussions on the antibiotic crisis were published over the past two years. These include articles on the antibiotic crisis18, 19, 20, 21, 22 and possible solutions,23, 24 sources of resistance,25 surveillance of resistance,26 restricting non-medical antibiotic use,27 antibiotic resistance in livestock and the environment,28, 29 possible approaches to address R&D and commercialization challenges,30, 31, 32 difficulties in discovering new antibiotics,33 development of new antibiotics,34, 35, 36 reviews of therapeutic strategies37 and new approaches to discover novel antimicrobials38, 39 to combat antibiotic resistance.
This review is an update of our 20112 and 20133 reviews and details antibiotics launched recently (Table 1; Figures 1, 2 and 3), as well as the development status, mode of action, spectra of activity, historical discovery and origin of the drug pharmacophore (natural product (NP), NP-derived, synthetic (S) or protein/mammalian peptide (P)) of antibiotics and β-lactam/β-lactamase inhibitor combinations undergoing clinical development (phase-I, -II or -III trials and regulatory evaluation) as of December 2015 (Tables 2, 3, 4 and 5; Figures 2, 3, 4, 5, 6, 7, 8, 9, 10). Compounds for which no development activity has been reported since our 2013 review3 are listed in Table 6. The ClinicalTrials.gov NCT codes are listed in parentheses for each trial and trials not in this database are referenced. New trials of approved drugs including new formulations are not discussed in this review. Data in this review were obtained by analyzing the journal literature and internet resources such as company web pages, clinical trial registers and biotechnology-related newsletters. Although every effort has been undertaken to ensure that this data is accurate, it is possible compounds undergoing early clinical development with limited information in the public domain have been overlooked. An overview of the drug development and approval process, antibiotic clinical trial categories and abbreviations are found in the Supplementary Information.

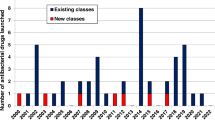
New antibiotic approvals 2000–2015 with new classes highlighted.

Structures of the recently launched antibiotics, perchlozone (1), the 1950s TB drug thiacetazone (13), delamanid (2), dalbavancin (3), oritavancin (4), tedizolid phosphate (5), nemonoxacin (8), finafloxacin (9) and ozenoxacin (12).

Structures of the recently lauched antibiotics β-lactam/β-lactamase inhibitor combinations ceftolozane (6)/tazobactam (7) and ceftazidime (10)/avibactam (11).

Structures of NP-derived compounds in phase-III clinical trials.

Structures of synthetic compounds in phase-III clinical trials.

Structures of NP-derived and P-derived compounds in phase-II clinical trials.

Structures of synthetic compounds in phase-II clinical trials.

Structures of compounds in phase-I clinical trials.

Structure of the β-lactamase inhibitor and β-lactam antibiotic combinations in phase-III clinical trials.

Structures of β-lactamase inhibitors and associated β-lactam antibiotics in phase-I clinical trials.
Antibacterial drugs launched since 2000
Since 2000, 30 new antibiotics (two NP, 12 NP-derived and 16 synthetic-derived) and two new β-lactam/β-lactamase inhibitor combinations have been launched worldwide (Table 1). Of the 30 new antibiotics, five were first-in-class antibiotics: linezolid (oxazolidinone, S, 2000), daptomycin (lipopeptide, NP, 2003), retapamulin (pleuromutilin, NP-derived, 2007), fidaxomicin (tiacumicin, NP, 2011) and bedaquiline (diarylquinoline, S, 2012). Importantly, these five new antibiotic classes only target Gram-positive (G+ve) bacteria, which reiterate the importance of identifying new antibiotic classes with Gram-negative (G−ve) activity. There was one new diazabicyclooctane (DBO)-type β-lactamase inhibitor (avibactam, S, 2015), which when used in combination with selected β-lactams also displays activity against G−ve bacteria.
There has been a steady launch of antibiotics since 2000, averaging three approvals every two years (Figure 1), with a notable spike of seven approvals in 2014. Of the 16 synthetically-derived new antibiotics, eleven were quinolones, two were oxazolidinones, and the remainders were single examples of the nitroimidazole, thiosemicarbazone and diarylquinoline classes. The β-lactam and glycopeptide classes accounted for six and three of the two NP and 12 NP-derived antibiotics respectively with the other five belonging to separate classes.
Since the last review,3 seven new antibiotics and two new β-lactam/β-lactamase inhibitor combinations (Figure 2) have been approved. These are discussed in detail below, along with perchlozone (1) that was approved in Russia in late 2012.
Thioureidoiminomethylpyridinium perchlorate (1) (Perchlozone) is an oral treatment developed by JSC Pharmasyntez (Moscow, Russia) and approved in Russia in 2012 for the treatment of multidrug-resistant tuberculosis. Perchlozone (1) was synthesized40 at the Siberian Division of the Russian Academy of Sciences (Novosibirsk, Russia) and is the perchlorate salt of thioureidoiminomethylpyridine, which was first reported in the early 1950s.41, 42 Perchlozone (1) is an analog of thiacetazone (13), a tuberculosis (TB) drug developed in the 1950s that fell out of favor due to adverse side effects.43 A recent study has proposed that 1 and thiacetazone (13) share a common mode of action: activation by the monooxygenase EthA and inhibition of the FASII dehydratase complex HadABC leading to disruption of mycolic acid biosynthesis.44
Delamanid (2) (Deltyba, OPC-67683) was developed by the Otsuka Pharmaceutical Co. (Tokyo, Japan) and was approved in Europe and Japan in April and July 2014 respectively as part of an appropriate combination regimen in adult patients with pulmonary multidrug-resistant (MDR)-TB on the basis of phase-IIb data.45, 46 Delamanid (2) is currently being evaluated in a phase-III trial for the treatment of multidrug-resistant TB in combination with other TB and retroviral drugs over six months (NCT01424670). Delamanid (2)47, 48 is derived from the anti-TB lead, bicyclic nitroimidazole CGI-17341,49, 50 which was dropped from development due to mutagenicity concerns. Delamanid (2) is a prodrug that is reductively activated by a deazaflavin (F420) dependent nitroreductase and inhibits mycobacterial growth through inhibition of mycolic acid biosynthesis.50
Dalbavancin (3) (Dalvance, Xydalba, BI-397) is one of two new glycopeptides that received FDA approval in 2014 after decades of development.51, 52, 53 It is a semi-synthetic lipopeptide analog of the teicoplanin-like glycopeptide A40926 Factor B, modified by amidation of the C-terminal acid group with N,N-dimethyl-1,3-diaminopropane. Originally developed at the Lepitit Research Centre of Marion Merrell Dow, it passed through the hands of Hoechst, Biosearch Italia S.p.A, Vicuron Pharmaceuticals, Pfizer (Groton, CT, USA), and finally Durata Therapeutics, who acquired the program in 2009. Durata conducted the final phase-III trials for acute bacterial skin and skin structure infections (ABSSSI) that led to FDA approval in May 2014 for treatment of G+ve ABSSI infections in adult patients caused by susceptible isolates of Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysagalactiae, Streptococcus anginosus group (including S. anginosus, S. intermedius, and S. constellatus) and E. faecalis (vancomycin-susceptible strains). Durata was acquired by Actavis in November 2014, and Actavis by Allergan, plc (Dublin, Ireland) in March 2015. Dalbavancin (3) has an extended half-life of over 300 h in humans,54 allowing for a significant dosing advantage over the twice-daily dosing required for vancomycin. The current prescribing information (Jan 2016) provides for two dosage regimes with different doses depending on renal function: if estimated creatinine clearance (CrCl) is ⩾30 ml min−1 (or on hemodialysis) then a single dose of 1500 mg or a two-dose regimen of 1000 mg followed by 500 mg is recommended. If CrCl is <30 ml min−1, then either a single dose of 1125 or a two-dose regimen of 750 mg followed by 375 mg is recommended. The EU Commission granted marketing authorization for dalbavancin (3) in March 2015, with its commercialization partnered with Angelini (Rome, Italy).55 In October 2015 Allergan filed a supplemental New Drug Application to expand the label to include a single 1500 mg dose administration,56 based on a phase-III trial in 698 patients (NCT02127970).57 Like other glycopeptides, dalbavancin (3) blocks peptidoglycan synthesis by binding to the precursor Lipid II, though additional mechanisms are proposed to help account for its increased potency.52, 58
Oritavancin (4) (Orbactiv, LY333328), the second glycopeptide achieving US Food and Drug Administration (FDA) approval in 2014, also navigated a tortuous route to market: initial development by Eli Lilly (Indianapolis, IN, USA) in the 1990s was followed by ownership by InterMune, Inc. (Brisbane, CA, USA), Targanta Therapeutics, and The Medicines Company (Parsippany, NJ, USA), who completed two additional phase-III trials and achieved FDA approval in August 2014.59 Oritavancin (4) is a semi-synthetic derivative of the vancomycin-like glycopeptide chloroeremomycin, which is alkylated on the vancosamine amine with a hydrophobic chlorophenyl-benzyl moiety. Compared to vancomycin it also possesses an additional aminosugar substituent. Like dalbavancin (3), an extended >300 h half-life60 allows for treatment of G+ve ABSSSI in adults with S. aureus (including methicillin-susceptible and methicillin-resistant isolates), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus anginosus group (includes S. anginosus, S. intermedius, and S. constellatus), or E. faecalis (vancomycin-susceptible isolates only) with a single 1200 mg dose infused over 3 h. Oritavancin (4) is designated as a Qualified Infectious Disease Product (QIDP) under the USA Generating Antibiotic Incentives Now (GAIN) act, providing a five-year extension of non-patent exclusivity. The European Medicines Agency (EMA) provided marketing authorization for oritavancin (4) in the 31 countries of the European Economic Area in March 2015.61 Multiple mechanisms of action, in addition to Lipid II binding, are proposed to increase the effectiveness of 4.62, 63, 64, 65, 66, 67
Tedizolid phosphate (5) (Sivextro, torezolid phosphate, TR-701, DA-7218) is an oxazolidinone prodrug that is dephosphorylated in vivo. It was approved by the FDA in June 2014 and in EU in June 2015 for the treatment of G+ve ABSSSI,68, 69, 70 and is currently in a phase-III trial for the treatment of presumed G+ve hospital-acquired bacterial pneumonia (HABP) or ventilator-associated bacterial pneumonia (VABP) (NCT02019420). Importantly, tedizolid phosphate (5) has once-daily dosing using either IV or oral administration and is active against linezolid-resistant strains.71 Tedizolid was discovered72 by Dong-A Pharmaceutical (Seoul, South Korea) with later development undertaken by Trius Therapeutics, acquired by Cubist Pharmaceuticals in January 2015, who in turn were acquired by Merck & Co. (Rahway, NJ, USA).73 Phase-III development has also been undertaken by Bayer Healthcare in Latin American countries and the Asia Pacific Region. Tedizolid (5) inhibits bacterial protein synthesis through binding to the 50S ribosome, preventing the formation of the 70S initiation complex.74
Zerbaxa (CXA-201)75 is an IV-administered combination of the cephalosporin ceftolozane (6) (CXA-101, FR264205) and the β-lactamase inhibitor tazobactam (7) that was approved by the FDA in December 2014 and the EMA in September 2015 for the treatment of complicated intra-abdominal infections (cIAI) in combination with metronidazole,76 and complicated urinary tract infections (cUTI) including pyelonephritis (kidney infection).77 Ceftolozane (6) is a fifth generation cephalosporin that displays broad-spectrum G−ve activity with potent activity against Pseudomonas aeruginosa,78, 79, 80 whereas tazobactam (7) is a β-lactamase inhibitor first approved in 1992 in combination with piperacillin.81, 82 Ceftolozane (6) was discovered by Astellas (Tokyo, Japan) and late stage clinical development was undertaken by Cubist, now Merck & Co. The combination of ceftolozane and tazobactam is currently under evaluation for the treatment of adult patients with either VABP or HABP (NCT02070757).
Nemonoxacin (8) (Taigexyn, TG-873870)83, 84 is a non-fluorinated quinolone from TaiGen Biotechnology Co., Ltd (Taipei, Taiwan) that had an oral formulation approved in Taiwan in March 2014 for the treatment of community-acquired bacterial pneumonia (CABP). In August 2015, TaiGen announced that their partner in China, Zhejiang Medicine Company (Hangzhou, China) had completed an on-site inspection by the Chinese FDA with an approval decision pending.85 TaiGen licensed 8 in 2005 from Procter & Gamble Pharmaceuticals, who were later acquired by Warner Chilcott, now incorporated into Allergan plc (Dublin, Ireland). Nemonoxacin (8) has broad-spectrum activity against resistant G+ve and G−ve strains;86, 87, 88 a phase-III trial using IV administration has completed recruitment (NCT02205112), whereas a phase-II trial to treat diabetic foot infections was completed in late 2014 (NCT00685698).
An otic solution of finafloxacin (9) (Xtoro, BAY 35–3377) developed by Alcon Laboratories (Fort Worth, TX, USA) was approved by the FDA in December 2014 to treat acute otitis externa, which is commonly known as ‘swimmer’s ear’, caused by P. aeruginosa and S. aureus infections.89 Alcon licensed finafloxacin (9) from MerLion Pharmaceuticals (Singapore, Singapore), who recently announced that finafloxacin was more efficacious than ciprofloxacin in phase-II trial for the treatment of cUTI (NCT01928433).90 Finafloxacin (9) is differentiated from other quinolones due to its improved activity at slightly acidic pH, which is more representative of physiological conditions.91, 92, 93, 94
Avycaz (CAZ104, CAZ-AVI) is a combination of the third-generation cephalosporin ceftazidime (10)95 and the new DBO-type β-lactamase inhibito r avibactam (11)96, 97, 98 that has in vitro activity against Enterobacteriaceae in the presence of some β-lactamases and extended-spectrum β-lactamases (ESBLs) including AmpC, TEM, SHV, CTX-M, Klebsiella pneumoniae carbapenemase (KPCs), AmpC, and certain oxacillinases (OXA), though it is not active against bacteria that produce metallo-β-lactamases or that overexpress efflux pumps. The combination restored the activity of ceftazidime in animal models of infection caused by ESBL, KPC and AmpC producing bacteria. It was approved by the FDA in February 2015 for the treatment of cIAI in combination with metronidazole, and cUTI including pyelonephritis. Actavis (formerly Forest, now Allergan) received this approval in collaboration with AstraZeneca (London, UK). Notably, this β-lactam and β-lactam inhibitor combination was the first antibiotic regimen to be approved by the FDA based only on phase-II data from 169 adult patients. In light of the limited clinical data Avycaz has a label restriction: ‘As only limited clinical safety and efficacy data for Avycaz are available, reserve Avycaz for use in patients who have limited or no treatment options.’ Two further phase-II trials are testing the combination in children (NCT02497781 and NCT02475733), whereas multiple phase-III trial have recently been completed using ceftazidime-avibactam in hospitalized adults with nosocomial pneumonia (NCT01808092), for cUTI (NCT01644643, NCT01595438, NCT01599806), or for cIAI (NCT01500239, NCT01499290, NCT015002389). In February 2016 Allergan announced the FDA has accepted for filing the company's supplemental New Drug Application (NDA) for Avycaz, adding new clinical data to the current label from two of the phase-III trials that evaluated Avycaz, in combination with metronidazole, for the treatment of cIAI, including patients with infections due to ceftazidime-non-susceptible pathogens. The FDA granted priority review status due to a QIDP designation for Avycaz. Avibactam (11) has also been evaluated in a phase-II trial (NCT01281462) in combination with ceftaroline fosamil and a phase-I trial with aztreonam (54) (NCT01689207) (Table 4; Figure 11).

Compounds under clinical evaluation divided into development phases and their lead derivation source (natural product (NP), synthetic (S), protein/peptide (P)) with the β-lactam/β-lactamase inhibitor (BLA) combinations listed separately.
Ozenoxacin (12) (Zebiax, M5120, T-3912), which was discovered by Toyama Chemical (Tokyo, Japan) and later developed by Maruho Co., Ltd (Osaka, Japan) was approved in Japan in September 2015 for the treatment of superficial S. aureus and S. epidermidis skin infections and acne that is accompanied by purulent inflammation.99 Ozenoxacin (12) is a non-fluorinated quinolone with broad-spectrum activity against a variety of susceptible and quinolone-resistant bacteria.100, 101 Ozenoxacin (12) is also being developed in Europe by Ferrer Internacional S.A. (Barcelona, Spain), who has licensed the USA rights to Medimetriks Pharmaceuticals, Inc. (Fairfield, NJ, USA)102 and the Canadian rights to Cipher Pharmaceuticals (Mississauga, ON, Canada).103 Ozenoxacin (12) has completed a phase-III trial (NCT01397461) and is currently undergoing another phase-III trial (NCT02090764) for the treatment of patients with impetigo.
Compounds undergoing clinical evaluation
The compounds currently undergoing clinical trials or under regulatory evaluation for the treatment of bacterial infections as of the end of December 2015 are detailed in the following tables and figures: phase-III in Tables 2 and 5 with structures in Figures 4,5 and 9, phase-II in Table 3 with structures in Figures 6 and 7, and phase-I in Tables 4 and 6 with structures in Figures 8 and 10.
NP and NP-derived compounds in phase-III trials
Solithromycin (14) (CEM-101) is a semi-synthetic 2-fluoroketolide discovered by Optimer that is being developed by Cempra Pharmaceuticals (Chapel Hill, NC, USA). It recently completed two phase-III trials for the treatment of CABP (NCT01968733 and NCT01756339). Solithromycin (14) demonstrated non-inferiority to moxifloxacin within 72 h, meeting the FDA’s primary endpoint, but was inferior in a 5–10 day follow-up required by the EMA. Furthermore, 34% of patients reported adverse events (mainly infusion site related), compared to 13% for moxifloxacin.104 Solithromycin (14) is a protein synthesis inhibitor105 that has broad-spectrum antibacterial activity including many ketolide/macrolide resistant strains including Neisseria gonorrhoeae, Chlamydia trachomatis and Mycoplasma genitalium.106, 107, 108, 109, 110 Solithromycin (14) is also being investigated in a phase-III trial for the treatment of uncomplicated urogenital gonorrhea (NCT02210325) and as an anti-inflammatory111 in a phase-II trial for the treatment of nonalcoholic steatohepatitis (NCT02510599). BARDA awarded Cempra a contract in 2013 to develop 14 in pediatric populations and for bioterror threat pathogens.112, 113 Toyama Chemical Co., Ltd (Tokyo, Japan) is also evaluating 14 (coded T-4288) in a phase-II trial in Japan.
Omadacycline (15) (amadacycline, PTK-0796) is a semi-synthetic minocycline derivative114 developed by Paratek Pharmaceuticals (Boston, MA, USA) with broad-spectrum antibacterial activity115 that can be administered either orally or IV. Omadacycline (15) recently started new phase-III trials for the treatment of CABP (NCT02531438) and ABSSSI (NCT02378480). Omadacycline (15) inhibits protein synthesis through enhanced ribosome binding compared to tetracycline at a similar site and maintains activity in the presence of the ribosomal protection proteins such as Tet(O) and Tet(M).116
Sarecycline (16) (P005672, PTK-AR01) is a semi-synthetic tetracycline derivative117 discovered by Paratek Pharmaceuticals (Boston, MA, USA) and licensed to Warner Chilcott in July 2007, who were acquired by Actavis now Allergan, plc (Dublin, Ireland) in October 2013. Sarecycline (16) is being evaluated in three phase-III trials for the treatment of acne vulgaris (NCT02413346, NCT02320149 and NCT02322866).
Eravacycline (17) (TP-434) is a synthetic fluorocycline-type tetracycline derivative118, 119, 120 developed by Tetraphase Pharmaceutics (Watertown, MA, USA) with broad-spectrum activity, including activity against bacteria that have acquired tetracycline-specific efflux and ribosomal protection through inhibition of protein synthesis.121, 122, 123 Eravacycline (17) was evaluated in phase-III trials for the treatment of cIAI (NCT01844856) and cUTI (NCT01978938). In December 2014 tetraphase announced that the IGNITE1 trial in cIAI met its primary endpoint, demonstrating high cure rates in prevalent G−ve pathogens and a favorable safety profile,124 but in September 2015 the top-line results for the cUTI trial showed 17 did not achieve either the FDA or EMA primary endpoints of statistical non-inferiority compared to levofloxacin.125 Eravacycline (17) was awarded QIDP designation for cUTI and cIAI in 2013.126
Surotomycin (18) (MK-4261, CB-183,315) is a semi-synthetic daptomycin derivative127, 128 developed by Cubist Pharmaceuticals, which was acquired by Merck & Co. (Rahway, NJ, USA) in December 2014, that completed two phase-III trials (NCT01597505 and NCT01598311) in 2015 for the treatment of Clostridium difficile infections (CDI). Surotomycin (18) has improved activity against C. difficile strains compared to daptomycin,129, 130, 131, 132 and against daptomycin-resistant S. aureus, E. faecalis and E. faecium.127 Importantly, it was recently reported that surotomycin (18) had only a modest disruptive effect on the gut microbiota in a phase-I trial, which could help reduce infection recurrences.133
Plazomicin (19) (ACHN-490)134, 135 is a semi-synthetic derivative136 of the aminoglycoside sisomicin137, 138 that has broad-spectrum activity against both G+ve and G−ve bacteria.139, 140, 141, 142 Plazomicin (19) is being evaluated by Achaogen, Inc. (South San Francisco, CA, USA) in phase-III trials for the treatment of cUTI and pyelonephritis (NCT02486627) and carbapenem-resistant Enterobacteriaceae VAP, HAP and bloodstream infections (NCT01970371).
Cefilavancin (20) (RD-1792, TD-1792) is a cephalosporin/glycopeptide heterodimeric antibiotic developed by Theravance Biopharma, Inc. (South San Francisco, CA, USA) that was licensed to R-Pharm (Moscow, Russia) in October 2012. In March 2015, R-Pharm reported that cefilavancin (20) was undergoing a phase-III trial as a treatment of G+ve complicated skin and skin structure infections with data expected in 2016.143 Cefilavancin (20) had previously completed a phase-II trial in 2007 that evaluated two mg/kg/day IV dosing versus vancomycin in 197 patients (NCT00442832), with similar efficacy.144 Cefilavancin (20) consists of vancomycin functionalized with a linker attached to the C-terminal carboxyl group via an amide bond; the other end of the linker is attached through an oxime linkage to the cephalosporin lactam amine substituent, resulting in a hybrid with a dual targeting action against peptidoglycan synthesis.145
Synthetic compounds in phase-III trials
SQ 109 (21), which was discovered by the National Institute of Allergy and Infectious Diseases (NIAID)146 and initially developed by Sequella, Inc. (Rockville, MD, USA), is an ethambutol analog that Infectex (Moscow, Russia) is evaluating in a phase-II/III trial for the treatment of MDR pulmonary TB.147 Recent work has shown that the mode of action of SQ 109 (21) is likely to be via dissipation of the transmembrane electrochemical proton gradient.148 Interestingly, SQ 109 (21) has shown potent killing activity of Trypanosoma cruzi, which is the parasite that causes Chagas’ disease.149, 150
Cadazolid (22) (ACT-179811) is a quinolonyl-oxazolidinone chimeric antibiotic under development by Actelion Pharmaceuticals (Basel, Switzerland) that is being evaluated in two phase-III trials for the treatment of patients with CDI (NCT01983683 and NCT01987895). The FDA has designated cadazolid (22) as a Fast Track development program and as a QIDP. Cadazolid (22) is a potent inhibitor of C. difficile protein synthesis,151 and also strongly suppresses toxin and spore formation.152, 153
Pretomanid (23) (PA-824), a nitroimidazole derivative being tested by the Global Alliance for TB Drug Development (New York, NY, USA), is in phase-III trials in combination with linezolid (NCT02333799), moxifloxacin and pyrazinamide (NCT02342886) and a multidrug regimen of bedaquiline, moxifloxacin, linezolid and clofazimine (NCT02589782). Pretomanid (23) is an analog of CGI-17341, which was found to be too toxic for clinical development,154 and is a prodrug that is reductively activated by the deazaflavin (cofactor F420)-dependent nitroreductase Rv3547. Under aerobic conditions 23 inhibits cell wall growth by hindering mycolic acid formation and under anaerobic conditions its activity involves the induction of respiratory poisoning.155, 156, 157
Delafloxacin (24) (RX-3341, WQ-3034, ABT-492)158 is being evaluated by Melinta Therapeutics (New Haven, CT, USA) and has completed one phase-III trial (NCT01811732) and is undergoing another (NCT01984684), both for the treatment of G+ve and G−ve ABSSSI. A phase-III trial for the treatment of uncomplicated gonorrhea was terminated (NCT02015637). In the completed ABSSSI study, delafloxacin (24) met the study’s primary endpoint, reduction in lesion size by at least 20% at 48–72 h in the intent-to-treat population without non-study antibiotics or major procedures, which was comparable to the response in the control arm receiving vancomycin plus aztreonam (54).159 Delafloxacin (24) has been assigned QIDP status by the FDA for this therapeutic area, as well as for the treatment of CABP.158 Delafloxacin (24), like finafloxacin (9), displays enhanced activity against G+ve bacteria at pH 5 due to its slightly anionic characteristics.160 The quinolone antibiotic class kill bacteria using a dual mechanism of DNA gyrase (GyrA) and topoisomerase IV (ParC) inhibition, with the GyrA/ParC activity ratios depending on the compound and microorganism target.161
Lascufloxacin (25) (KRP-AM1977X)162 is an orally administered quinolone developed by Kyorin Pharmaceutical Co., Ltd (Tokyo, Japan) that entered phase-III trials in Japan in April 2015 for the treatment of respiratory infections.163 An IV formulation of 25 called KRP-AM1977Y is currently in phase-II trials.163
Zabofloxacin (26) (PB-101, DW-224a), which is being developed Dong Wha Pharmaceutical (Seoul, South Korea), completed a phase-III trial in late 2014 for the treatment of patients with acute bacterial exacerbation of chronic obstructive pulmonary disease (NCT01658020).164 There has been no further update from Dong Wha on the development status of 26.
NP and NP-derived compounds in phase-II trials
Exeporfinium chloride (27) (XF-73) is a porphyrin derivative being developed by Destiny Pharma (Brighton, UK) that has been evaluated in phase-I/II trials for the prevention of post-surgical staphylococcal nasal infections (NCT02282605).165 Exeporfinium chloride (27) is a photosensitizer that has broad-spectrum G+ve activity166, 167, 168, 169, 170 and activity against Candida albicans.171
Auriclosene (28) (NVC-422, N,N-dichloro-2,2-dimethyltaurine) is an N-dichlorotaurine analog being evaluated in phase-II trials by NovaBay Pharmaceuticals, Inc. (Emeryville, CA, USA) as an irrigation solution on urinary catheter patency (NCT02130518). It recently completed a phase-II trial for the treatment of bacterial conjunctivitis (NCT01877694). Auriclosene (28) was designed to be a more stable derivative of the naturally occurring oxidant N-dichlorotaurine172, 173, 174, 175 and also was recently shown to inactivate S. aureus toxins.176
Lefamulin (29) (BC-3781) is a semi-synthetic pleuromutilin177, 178 derivative originally discovered by Nabriva Therapeutics AG (Vienna, Austria). Nabriva executed an Initial Public Offering of shares in September 2015, raising $92 m to progress lefamulin (29) into phase-III trials for CABP.179 The first, NCT02559310, is currently recruiting 738 patients for a comparison with moxifloxacin +/− linezolid, using IV 29 with potential step-down to oral 29. A phase-II trial for ABSSSI was completed in 2012 (NCT01119105),180 whereas further trials in ABSSSI, HABP and VABP are planned.179 Lefamulin (29) is a protein synthesis inhibitor that displays antibacterial activity against a range of skin and respiratory pathogens.177, 181, 182
CB-06-01 (30) (NAI-003, BIK-0376, NAI-Acne) is an amide derivative of GE2270, a cyclic thiazole peptide obtained from fermentation of a Planobispora rosea strain that is active against G+ve bacteria and anaerobes.183 CB-06-01 (30), licensed from NAICONS (Milan, Italy), is being developed by Cassiopea S.p.A. (Milan, Italy) (formerly Cosmos S.p.A.) as a topical treatment for acne infections. According to the company website, 30 has completed a phase-I study and is currently undergoing phase-III POC trials, expected to be completed in 2Q 2016, though no trials are listed with ClinicalTrials.gov.183
Cefiderocol (31) (S-649266, GSK-2696266) is a chimeric cephalosporin with a catechol siderophore substituent184, 185, 186 from Shionogi & Co., Ltd (Osaka, Japan) being co-developed with GSK (London, UK). It completed phase-I testing187, 188, 189 and is currently being assessed in a phase-II trial (NCT02321800) for cUTIs caused by G−ve pathogens in hospitalized adults in comparison with IV imipenem/cilastatin.
Protein/mammalian peptide-derived compounds in phase-II trials
POL7080 (RG7929) is a synthetic cyclic peptide based on protegrin I,190, 191 which was first isolated from porcine leucocytes,192 but the structure has yet to be made publically available. Developed by Polyphor, Ltd (Basel, Switzerland), POL7080 successfully completed a phase-I trial193, 194 and was partnered with Roche in 2013.195 POL7080 completed a phase-II trial in 20 patients with exacerbation of non-cystic fibrosis bronchiectasis in November 2015 (NCT02096315) and is currently undergoing another phase-II study in 25 patients with P. aeruginosa VABP co-administered with standard of care (NCT02096328). The collaboration with Roche was discontinued in November 2015,196 with Polyphor continuing the phase-II trial on its own. POL7080 has potent and selective antimicrobial activity against G−ve bacteria including P. aeruginosa and has a novel mode of action through targeting the β-barrel protein LptD (Imp/OstA), which is involved in the outer-membrane biogenesis of lipopolysaccharide.190, 194
Brilacidin (32) (PMX-30063), a membrane targeting arylamide oligomer licensed from the University of Pennsylvania that was being developed by Polymedix Inc. (Radnor, PA, USA), completed a phase-IIa trial for the treatment of ABSSSI in 2012 (NCT01211470). The future development of brilacidin (32) appeared in doubt as Polymedix filed for bankruptcy on 1 April 2013 and there were reports of possible toxicity concerns,197 but the assets of Polymedix were acquired by Cellceutix (Beverly, MA, USA) in September 2013 for $US2.1 m plus 1.4 m shares, an apparent bargain considering Polymedix had a market capitalization of over $200 m in 2012.198 Cellceutix conducted a phase-IIb trial in 2014 (NCT02052388), comparing 32 against daptomycin for treatment of ABSSSI in 215 patients, with no serious adverse events and efficacy similar to daptomycin across all brilacidin treatment groups, including the two single-dose groups.199 Brilacidin (32) received QIDP designation under the GAIN Act in November 2014.200 Another phase-II trial (NCT02324335) is currently recruiting to test an oral rinse of brilacidin to treat oral mucositis in cancer patients.201 Cellceutix announced in 2015 that 32 would proceed to a phase-III trial.202 Brilacidin (32) is a member of the family of arylamide foldamers that was designed to mimic cationic antimicrobial peptides and had shown bactericidal activity against both G+ve and G−ve bacteria.203, 204, 205 Cellceutix is also working on a defensin mimetic-compound CTIX-1278 that is still in preclinical testing.206
LTX-109 (33) (Lytixar), a cationic peptide mimic,207 is being developed by Lytix Biopharma AS (Oslo, Norway) as a topical antimicrobial peptide. It completed phase-II trials for the treatment of impetigo (NCT01803035) in April 2014 and uSSSI (NCT01223222) in February 2011, and a phase-I/II trial for nasal decolonization of S. aureus including MRSA (NCT01158235).208 LTX-109 (33) has rapid bactericidal in vitro activity against both G+ve and G−ve drug-resistant strains.209, 210 Lytix planned to develop 33 through a phase-I/II trial for the treatment of mild diabetic foot infections, but in October 2015 announced it had decided not to proceed with the program due to higher costs, a longer than anticipated timeframe, and the company’s focus on oncology.211
Synthetic compounds in phase-II trials
Radezolid (34) is an ‘enhanced’ oxazolidinone antibiotic, developed by Melinta Therapeutics, Inc. (New Haven, CT, USA), which has activity against G+ve bacteria (including those with resistance to linezolid) in addition to some G−ve bacteria such as Haemophilus influenzae and Moraxella catarrhalis.212 Radezolid (34) was rationally designed from the overlap of sparsomycin and linezolid binding sites on the 50S ribosomal subunit.213, 214, 215 Radezolid (34) completed two phase-II trials for CABP (NCT00640926, 2009) and uSSSI (NCT00646958, 2008), but no further development has been reported. In January 2015 Melinta licensed radezolid to Malin Corporation plc (Dublin, Ireland) for topical uses.216
MRX-I (35) is a new oxazolidinone antibiotic being developed by MicuRx (Hayward, CA, USA and Shanghai, China), proposed to possess an improved safety profile compared to linezolid.217, 218, 219 It completed a phase-I trial in April 2012,220 and is currently undergoing testing in a phase-II comparison with linezolid for the treatment of ABSSSI (NCT02269319: 120 patients, January-October 2015).
Ridinilazole (36) (SMT19969) is a synthetic bibenzimidazole compound221 from Summit Corporation PLC (Oxford, UK) that is being developed for the treatment of CDI in collaboration with the Wellcome Trust (London, UK). Ridinilazole (36) completed a phase-I study that showed 36 was tolerated at therapeutically relevant doses and was highly sparing of gut flora, with only the clostridia bacterial family being reduced to levels below the limit of detection.222 The Wellcome Trust awarded Summit an additional Translational Award for further development,222 leading to a phase-II study comparing 36 with vancomycin in C. difficile-associated diarrhea treatment that was initiated in April 2014, with a primary completion in November 2015 (NCT02092935). Positive top-line results were achieved, showing statistical superiority in sustained clinical response rates compared to the standard of care, vancomycin.223 Ridinilazole (36) has already received QIDP designation and has been granted Fast Track status from the US FDA,224 with phase-III trials being considered. Ridinilazole (36) showed good results in a hamster model of CDI.225
Gepotidacin (37) (GSK-2140944) is a bacterial Type II topoisomerase inhibitor226, 227 being investigated by GlaxoSmithKline (London, UK) that has completed multiple phase-I clinical trials (NCT01706315, NCT01615796, NCT01934205, NCT02000765, NCT02045849, NCT02202187 and NCT02257398), with both oral and IV dosing. A phase-II study investigating gepotidacin (37) with IV/oral switch therapy in G+ve ABSSSI was completed in June 2015 (NCT02045797), with a second phase-II study treating uncomplicated urogenital gonorrhea caused by N. gonorrhoeae initiated in April 2015 (NCT02294682).
ETX0914 (38) (AZD0914) is an orally available topoisomerase inhibitor developed by AstraZeneca (London, UK) that is in a phase-II clinical trial that was initiated in November 2014 (NCT02257918) to investigate efficacy in treating uncomplicated gonorrhea. It received FDA QIDP designation in June 2014,228 and formed one of the key assets of AstraZeneca’s anti-infective spinout company, Entasis Therapeutics (Waltham, MA, USA), in 2015.229 ETX0914 (38) is a novel spiropyrimidinetrione that targets bacterial topoisomerase II with a mode of inhibition distinct from the fluoroquinolones and aminocoumarins, meaning it retains activity against fluoroquinolone resistant isolates.230, 231
Debio-1452 (39) (AFN 1250) was being developed by Affinium Pharmaceuticals (Austin, TX, USA) and successfully completed a phase-IIa trial in August 2012 (NCT01519492) as an oral formulation for the treatment of staphylococcal ABSSSI. Debio-1452 (39) disrupts fatty acid biosynthesis by inhibiting staphylococcal FabI,232, 233 an essential enzyme in the final step of the fatty acid elongation cycle,234, 235 and was derived from a benzodiazepine identified at GSK (London, UK) using a high-throughput screen looking for inhibitors of S. aureus FabI.236, 237 Structure-based design led to the identification of a 3,4-dihydro-1,8-naphthyridin-2(1H)-one core with good in vitro and in vivo antibacterial activity with no significant cytotoxicity.236 Affinium licensed the program in 2002 and conducted further structure optimization leading to AFN-1252 (39).232, 238, 239, 240, 241, 242, 243, 244, 245 In February 2014, Debiopharm Group (Lausanne, Switzerland) acquired Affinium’s antibiotic assets, including both 39 and a prodrug of AFN-1252, Debio-1450 (AFN-1720).246 The prodrug, Debio-1450, was assessed in two phase-I studies (NCT02162199 and NCT02214433), with a third (NCT02595203) scheduled for completion in December 2015. A phase-II study was initiated in May 2015 that is examining IV switch oral therapy compared with IV vancomycin and oral linezolid in the treatment of patients with staphylococcal ABSSSI, estimated to be completed by June 2016 (NCT02426918). The FabI inhibitor platform is being used to generate other antibiotic candidates Debio 1453 and Debio 1454, for N. gonorrhoeae and Enteric species, respectively,247 in collaboration with Nobelex Biotech Inc. (Toronto, Canada), founded by Affinium’s former management.248
The fluoroquinolone WCK-771 (40), which is the arginine salt of S-(−)-nadifloxacin, and its prodrug alalevonadifloxacin (WCK-2349) (41)249 completed four phase-I trials between 2013 and August 2015 (NCT01875939, NCT02244827, NCT02217930, NCT02253342). Its developer, Wockhardt Limited (Mumbai, India) received QIDP status for WCK-771 (40) and alalevonadifloxacin (41) in August 2014,250 the first for an Indian company. Nadifloxacin is a racemic fluoroquinolone with a previous history of clinical use in Japan in 1993 as a topical antibiotic to treat acne and methicillin-resistant staphylococcal infections.251 The S enantiomer of nadifloxacin was found to be more active than the racemic mixture and had pharmacokinetic properties amenable for systemic use,251, 252, 253, 254 whereas the prodrug is used to provide oral availability.
Compounds in phase-I trials
BAL30072 (42)255, 256, 257 is a chimeric monobactam derivative with an iron-chelating dihydroxypyridone moiety developed by Basilea Pharmaceutica (Basel, Switzerland) for use as monotherapy or in combination with carbapenems. In 2013 Basilea was awarded a $US89 m contract by BARDA for the development of an IV dosage form,258, 259 leading to a phase-I study initiated in June 2014 to evaluate multiple-ascending doses of IV-administered 42 alone and in combination with meropenem (43).260 In September 2015, Basilea announced that an inhaled dosage form of 42 would be developed as part of the European iABC (inhaled Antibiotics in Bronchiectasis and Cystic fibrosis) program.261 This inhaled form of BAL30072 (42), a targeted treatment for chronic lung infections, will initially undergo preclinical development activities in preparation for phase-I studies.262 BAL30072 (42) displays activity against many G−ve bacteria255 including P. aeruginosa,263 Acinetobacter baumannii264, 265 and Burkholderia pseudomallei266 and is rapidly absorbed into bacteria via the essential iron uptake systems.255, 263 BAL30072 (42) was found to be effective against a series of clinical isolates from New York City hospitals.267
LCB01-0371 (44)268 is another new oxazolidinone, with a cyclic amidrazone substituent, belonging to LegoChem Biosciences, Inc. (Daejeon, South Korea). Purported to be more safe than linezolid or tedizolid, it has completed three phase-I trials (NCT01554995, NCT01842516 and NCT02540460) and has recently completed a phase-II trial (NCT01554995). Two additional phase-I trials are either currently recruiting (NCT02529241) or planned (NCT02538003).
TD-1607 (45), like cefilavancin (20) (TD-1792), is a heterodimeric antibiotic developed by Theravance Biopharma, Inc. (South San Francisco, CA, USA) consisting of vancomycin covalently linked to a cephalosporin, providing a dual targeting action against peptidoglycan synthesis. Although cefilavancin (20) forms the linkage with the C-terminal carboxyl group of vancomycin and an oxime group attached to the cephalosporin lactam amine substituent, TD-1607 (45) is aminomethylated on an aromatic ring (like telavancin) and this handle is used to attach the glycopeptide to the cephalosporin, which itself is attached via a pyridinyl substituent off the six-membered ring of the cephalosporin bicyclic ring system.269 TD-1607 (45) entered two phase-I trials, testing single IV dosing in 64 patients in April 2013 (NCT01791049), with the study completed in August 2013, and an additional 48 patients in September 2013-July 2014 (NCT01949103). No further development has been reported, though it is listed in the 2015 Theravance Biopharma annual report as ‘midstage’ candidate for MRSA.270
MGB-BP-3 (46) is a novel compound with G+ve activity being developed by MGB Biopharma (Glasgow, UK). It is based on the University of Strathclyde’s DNA minor groove binding technology271 and derived from distamycin A.272 A phase-I trial (NCT02518607) of 46 was initiated in July 2015 with an oral formulation being developed to treat CDI; IV and topical treatments are in preclinical development.273
CRS3123 (47) (REP-3123), a small molecule protein synthesis inhibitor that acts on the novel target methionyl-tRNA synthetase (MetRS) originally developed by Replidyne just under 10 years ago,274 is being developed by Crestone Inc. (Boulder, CO, USA) for the treatment of CDI. CRS3123 (47) is potent against a range of C. difficile clinical isolates, but has a relatively narrow spectrum of activity that may help to spare the normal gut bacteria during therapy. The first phase-I trial (NCT01551004), sponsored by the NIAID, enrolled 40 patients from May 2012-April 2014 to test a single dose. A second trial, also sponsored by NIAID, was initiated in April 2014 (NCT02106338) to test multiple doses in up to 30 subjects.275
TBA-354 (48)276 is a next-generation nitroimidazole being developed to treat TB by the TB Alliance, in conjunction with the University of Auckland and University of Illinois-Chicago.277 TBA-354 (48), shown to be more potent than pretomanid (23) in preclinical testing,278 is currently in two phase-I trials (NCT02288481 and NCT02606214), the first new TB drug candidate to begin a phase-I trial since 2009.279
Q203 (49) is a new imidazo[1,2-a]pyridine amide280, 281 with potent anti-tubercular activity that is being evaluated in a phase-I trial (NCT02530710) by Qurient Co., Ltd (Seongnam-si, South Korea). Qurient has also licensed 49 to Infectex (Moscow, Russia). Two imidazo[1,2-a]pyridine amides were identified from screening 121 526 compounds using a phenotypic high-content assay in infected macrophages.217 Structure optimization led to the identification of Q203 (49) that has excellent in vivo activity and pharmacokinetic and safety profiles compatible with once-daily dosing.217, 218 Q203 (49) and analogs inhibit Mycobacterium tuberculosis growth by targeting the respiratory cytochrome bc1 complex.217
β-Lactam/β-lactamase inhibitors undergoing clinical evaluation
The β-lactam antibiotics, which have evolved over multiple generations of improved subclasses, include penicillins, cephalosporins and carbapenems. They have been one of the most successful antibiotic classes ever discovered. Resistance to the β-lactams is generally caused by lactam ring opening by β-lactamase enzymes, inactivating the antibiotics so they are unable to inhibit their target, the bacterial penicillin-binding proteins involved in cell wall synthesis. This resistance can be overcome by co-administration of a β-lactamase inhibitor that provides the first clinical example of a ‘resistance-breaker’ strategy. The first β-lactamase inhibitor, clavulanic acid,282, 283, 284 was isolated from Streptomyces clavuligerus and is still used today as a combination therapy with amoxicillin, most commonly known as Augmentin. New β-lactam/β-lactamase inhibitor combinations were reviewed in 2013285, 286 and 2011,285, 286 whereas β-lactam antibiotics and β-lactamase inhibitors that are approved, in the clinic, and under development, were reviewed in 2014.287
β-Lactam/β-lactamase inhibitor combinations in phase-III trials
Carbavance is a combination of meropenem (43), a carbapenem first launched in Italy in 1994, and vaborbactam (RPX7009) (50), a novel boron-containing β-lactamase inhibitor,288 initially developed by Rempex Pharmaceuticals (San Diego, CA, USA), which was subsequently bought by The Medicines Company (Parsippany, NJ, USA) in 2013.289 Carbavance has completed multiple phase-I trials (NCT01702649, NCT01751269, NCT01772836, NCT02020434 and NCT02073812) and is currently recruiting two phase-III trials, comparing Carbavance to piperacillin+tazobactam in cUTIs (NCT02166476) and against best available therapy for carbapenem-resistant Enterobacteriaceae serious infections (cUTI, acute pyelonephritis, HABP, VABP, bacteremia; NCT02168946).
A combination therapy of imipenem (51), cilastatin (52) and relebactam (53) (MK-7655) that is being developed by Merck & Co. (Rahway, NJ, USA) has completed phase-II trials for cUTI (NCT01505634; completed July 2015) and cIAI (NCT01506271, completed August 2014). A phase-III trial comparing the combination against colistimethate sodium+imipenem+cilastatin in imipenem-resistant bacterial infection is currently recruiting 64 patients (NCT02452047) with an estimated completion in October 2016. A comparison with piperacillin+tazobactam for treatment of bacterial pneumonia in 536 patients is in preparation (NCT02493764), with an estimated completion in January 2018. Relebactam (53) is a DBO β-lactamase inhibitor related to avibactam (11),290 whereas imipenem (51) is a carbapenem first launched in 1987 that needs to be co-administered with the dehydropeptidase inhibitor cilastatin (52) in order to impede imipenem metabolism.291
β-Lactam/β-lactamase inhibitor combinations in phase-I trials
ATM-AVI292 is a combination of aztreonam (54), a monobactam first launched in 1984, and avibactam (11) (NXL104) that was being evaluated by AstraZeneca (London, UK) and Forest Laboratories (now part of Allergan, plc) in a phase-I trial (NCT01689207) that started in September 2012 but was not completed until December 2014. ATM-AVI was selected by the Innovative Medicines Initiative (IMI) ‘New Drugs for Bad Bugs’ (ND4BB) program in 2013. The IMI solicited proposals to evaluate the combination in a phase-IIa pharmacokinetic/pharmacodynamic analysis of ATM-AVI in patients with serious infections caused by G−ve pathogens, and in a phase-III randomized, multicenter, clinical study to evaluate the efficacy and safety of ATM-AVI in the treatment of serious infections caused by G−ve pathogens.293 AstraZeneca recently announced a new phase-II trial (NCT02655419) that will evaluate the PK, safety and tolerability of ATM-AVI for the treatment of cIAIs in hospitalized adults.
OP0595 (55) (RG6080) is another DBO β-lactamase inhibitor294 developed by Meiji Seika Pharma, Co. Ltd (Tokyo, Japan) and Fedora Pharmaceuticals (Edmonton, AB, Canada) and partnered with Roche in January 2015.295, 296 OP0595 (55) completed a phase-I trial (NCT02134834) in November 2014.
AAI101 is a novel extended-spectrum β-lactamase inhibitor developed by Allecra Therapeutics Gmbh (Weil am Rhein, Germany)/Allecra SAS (Saint Louis, France), an anti-infective company established in 2013 by Orchid Chemicals and Pharmaceuticals Ltd (Chennai, India) and lead investors Forbion Capital Partners (Naarden, The Netherlands) and Edmond de Rothschild Investment Partners (Paris, France).297, 298 Phase-I results were presented at ICACC 2014.299 Preclinical testing has examined the combination with cefepime (56).300, 301 Allecra has a second β-lactam antibiotic/β-lactamase inhibitor combination, AAI202 that has also completed phase-I trials,302 but no further information is available. No structures have been disclosed but they are likely to be a clavulanic acid-type inhibitors.298
Zidebactam (57) (WCK 5107)303 is a β-lactamase inhibitor being developed by Wockhardt Biopharm (Mumbai, India). It is being tested in a phase-I trial initiated in August 2015 alone and in combination with cefepime (56) (NCT02532140).
Compounds discontinued from clinical development
Analysis of compounds undergoing clinical trials
Numbers of compounds undergoing clinical evaluation and their source derivation
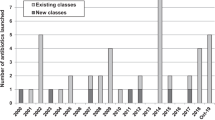
There are a total of 37 compounds and six β-lactam/β-lactamase inhibitor combinations currently undergoing clinical trials (Figure 11). Of the 37 compounds, 13 are in phase-III (Table 2; Figures 4 and 5), 17 in phase-II (Table 3; Figures 7 and 8) and seven in phase-I (Table 4; Figure 10), whereas there are two β-lactam/β-lactamase inhibitor combinations in phase-III and four in phase-I (Table 5; Figures 9 and 11). Nineteen antibiotics were synthetically-derived (S), 15 were NP-derived (NP), and three were protein/mammalian peptide-derived (P) (Figure 11). There is approximately the same number of compounds in each development phase in 2015 compared to 2013 (Figure 12). The only notable difference is between the number in phase-III trials in 2011 (6) compared to 2013 (16) and 2015 (15). Importantly, new antibiotics (ETX0914 (38), CB-06-01 (30), TD-1607 (45), CRS3123 (47; Replidyne had previously developed 47 as REP-3123 in phase-III trials in 2008), TBA-354 (48) and Q203 (49)) and β-lactamase inhibitors (OP0595 (55), AAI101 and zidebactam (57)) have entered the clinic to counteract those no longer being evaluated (Table 6).

New antibacterial pharmacophore analysis
The NP- and protein/mammalian peptide (P)-derived compound derivation is quite diverse but there are only four new antibacterial pharmacophores derived from NPs and three from P pharmacophores (Figure 13). The new NP and P pharmacophores are derived from porphyrin (phase-II; exeporfinium (27)/ porphyrin), N-chlorotaurine (auriclosene (28)/ N-chlorotaurine), thiopeptide (CB-06-01 (30)/ GE2270-A), and distamycin A (phase-I; MGB-BP-03 (46)), whereas brilacidin (32), LTX-109 (33) and POL7080 are derived from or inspired by naturally occurring cationic peptides. Similarly, the new synthetically-derived antibacterial pharmacophores are diverse: cadazolid (22; phase-III, oxazolidinone and quinolone chimera), ridinilazole (36; phase-II; bibenzimidazole), gepotidacin (37; phase-II, ‘gepotidacin’ class); ETX0914 (38; phase-II, spiropyrimidinetrione), Debio-1452 (39)/ Debio-1450 (both phase-II; benzodiazepine), CRS3123 (47; phase-I, diaryldiamine) and Q203 (49; phase-I, imidazo[1,2-a]pyridine amide). After the recent approval of the DBO class β-lactamase inhibitor avibactam (11) there is only one class of new pharmacophores: the boron class exemplified by vaborbactam (50), which is part of a combination with meropenem (43) called Carbavance. Although these classes have been classified as synthetically-derived for this review, they are inspired by the NP clavulanic acid, which was the first β-lactamase inhibitor reported.282, 283

Compounds [natural product (NP), synthetic (S), protein/peptide (P)] and β-lactamase (BLA) inhibitors with new antibacterial pharmacophores divided into development phases and their lead derivation source.
The total number of new pharmacophores rose from 11 in 2011 to 17 in 2013 and then back slightly to 15 in 2015 (Figure 14). Since 2013, five compounds with new antibacterial pharmacophores, LFF-571, lanopepden (GSK1322322), DPK-060 and ACHN-975 are not currently being actively pursued; whereas the peptide-derived LL-37 and IMX-942 have been retooled for anti-inflammatory purposes (Table 6).

Conclusion and future outlook
There was a steady state of compounds entering and leaving antibiotic clinical trials (Figure 12) and those with new pharmacophores (Figure 14) from 2013 to 2015. Of the 15 new pharmacophores, only a few have activity against G−ve bacteria: the topically-administered auriclosene (28) and LTX-109 (33), and the IV-administered POL7080, brilacidin (32), gepotidacin (36) and ETX0914 (38). Other non-quinolone G−ve actives are the aminoglycoside plazomicin (27), the β-lactam-siderophore hybrids S-649266 (31) and BAL30072 (44) and the six β-lactamase inhibitor/β-lactam combinations that includes vaborbactam (50), which is a novel boron-containing β-lactamase inhibitor in phase-III trials with meropenem (43). A worrying trend is that there is only one G−ve active, BAL30072 (44), in phase-I development, which started its first trial in 2010 and was re-booted in late 2015 to evaluate an inhaled dosage form. Hopefully, some of these compounds will move forward into phase-III trials so that new G−ve drugs will be available to physicians to treat MDR infections.
Unfortunately, as this review illustrates, the acute positive trend of new approvals masks a chronic underlying malaise in antibiotic discovery and development. While there are a number of antibiotics in late stage trials near approval, the pipeline feeding further New Drug Applications is drying up, with relatively few early stage candidates. There is still interest in antibiotics from large pharmaceutical companies, as evidenced by Roche partnering with several small biotech antibiotic companies in the past few years (Polyphor 2013—discontinued 2015196, RQx 2013, Spero Therapeutics 2014, Discuva Ltd 2014, GeneWeave 2015, Meiji Seika Pharma and Fedora 2015) and the Merck acquisition of Cubist for nearly $10 bn in 2014, although this is tempered by AstraZeneca shedding its antibiotic discovery group into Entasis Therapeutics (Waltham, MA, USA) in mid-2015. Many new antibiotics have arisen from smaller biotech companies, and this trend appears likely to continue with a number of new biotech startups forming such as Macrolide Pharmaceuticals (Watertown, MA, USA), Kaleido Biosciences (Cambridge, MA, USA) and Spero Therapeutics (Cambridge, MA, USA);304 there are nearly 40 European biotech companies in the BEAM (Biotechs from Europe innovating in Anti-Microbial Resistance; http://beam-alliance.eu) alliance.305
Antibiotics remain at the forefront of treating infections, but there is also a growing trend towards alternatives to antibiotics, which includes ‘non-compound’ approaches such as antibodies, probiotics, lysins, phage therapy, immune stimulation and vaccines,306 as well as a search for small molecule ‘resistance breakers’ designed to assist existing antibiotics overcome resistance,307 much as β-lactamase inhibitors help β-lactams retain activity. Attempts are being made to re-invigorate traditional antibiotic discovery by creating more effective ways to isolate novel natural products,308 such as assessing extremophiles or marine organisms grown under unusual conditions (for example, the Marine Bioproducts Engineering Center, Honolulu, HI, USA and Berkeley, CA, USA) or utilizing genomic screening technologies to look for specific sequences in gene expression libraries from DNA extracted from environmental samples that may be manipulated to produce novel antimicrobials309 (for example, Warp Drive Bio LLC, Cambridge, MA, USA). NovoBiotic Pharmaceuticals (Cambridge, MA, USA) is using an ‘iChip’ to culture and isolate bacteria in situ in soil, which led to the highly publicized discovery of teixobactin.310 Sanofi has partnered with the Fraunhofer Institute for Molecular Biology and Applied Ecology (Aachen, Germany) in a joint effort to explore natural products, mining Sanofi’s collection of >100 000 different microorganisms to cultivate them under various conditions and stimulate them to produce active substances.311 The Community for Open Antimicrobial Drug Discovery (Brisbane, Australia) is a Wellcome Trust and the University of Queensland initiative that is attempting to mine the chemical diversity contained in millions of organic chemist’s laboratories around the world to uncover new synthetic chemotypes.312, 313
The current clinical trial status of new antibiotics outlined in this review indicates we still face a serious threat from new extremely drug-resistant G−ve bacteria, including the polymyxin-resistant strains that generated a great deal of publicity in late 2015. There are few novel therapies in the pipeline, and innovative approaches for G−ve bacteria are scarce. The only light on the horizon is the continued increase in public and political awareness of the issue. Much of the innovation now appears to be driven by small biotech companies, with later stage partnerships with large pharma companies. However, with the continued retirement and retrenchment of experienced, qualified antibiotic development professionals, we potentially face a generational knowledge gap. It is now more important than ever to continue to search for and develop new antibacterial drug leads to stem a MDR bacteria tsunami that threatens to push us into a human health era where most G−ve infections will not be able to be treated. Equally importantly we need incentives to retain the few antibiotic researchers left today.
References
Cooper, M. A. & Shlaes, D. Fix the antibiotics pipeline. Nature 472, 32 (2011).
Butler, M. S. & Cooper, M. A. Antibiotics in the clinical pipeline in 2011. J. Antibiot. 64, 413–425 (2011).
Butler, M. S., Blaskovich, M. A. & Cooper, M. A. Antibiotics in the clinical pipeline in 2013. J. Antibiot. 66, 571–591 (2013).
Reardon, S. Antibiotic resistance sweeping developing world. Nature 509, 141–142 (2014).
World Health Organization. Antimicrobial resistance: global report on surveillance. https://apps.who.int/iris/bitstream/10665/112642/1/9789241564748_eng.pdf. (Accessed 28 December 2015).
Davies, S. C. Chief medical officer annual report, Vol 2. https://www.gov.uk/government/publications/chief-medical-officer-annual-report-volume-2 (Accessed 21 December 2015).
UK five year antimicrobial resistance strategy 2013 to 2018, September 2013. https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/244058/20130902_UK_5_year_AMR_strategy.pdf (Accessed 28 December 2015).
O'Neill, J. UK review on antimicrobial resistance. Tackling a global health crisis: initial steps, February 2015. http://amr-review.org/Publications (Accessed 28 December 2015).
Antibiotic resistance threats in the United States, 2013. U.S. Department of Health and Human Services (Centers for Disease Control and Prevention). http://www.cdc.gov/drugresistance/threat-report-2013 (Accessed 26 December 2015).
Report to the president on combating antibiotic resistance, September 2014. https://www.whitehouse.gov/sites/default/files/microsites/ostp/PCAST/pcast_carb_report_sept2014.pdf (Accessed 28 December 2015).
National Strategy for combating antibiotic-resistant bacteria. The White House, Washington, September 2014. https://www.whitehouse.gov/sites/default/files/docs/carb_national_strategy.pdf (Accessed 28 December 2015).
National Action Plan for combating antibiotic-resistant bacteria. The White House, Washington, March 2015. https://www.whitehouse.gov/sites/default/files/docs/national_action_plan_for_combating_antibotic-resistant_bacteria.pdf (Accessed 28 December 2015).
Erickson, B. E. Battling drug-resistant bacteria. Chem. Eng. News 92, 27–29 (2014).
Antimicrobial resistance and use in Canada A federal framework for action, Public Health Agency of Canada, October (2014).http://healthycanadians.gc.ca/alt/pdf/drugs-products-medicaments-produits/antibiotic-resistance-antibiotique/antimicrobial-framework-cadre-antimicrobiens-eng.pdf (Accessed 28 December 2015).
Federal action plan on antimicrobial resistance and use in Canada, Public Health Agency of Canada, March 2015. http://healthycanadians.gc.ca/alt/pdf/publications/drugs-products-medicaments-produits/antibiotic-resistance-antibiotique/action-plan-daction-eng.pdf (Accessed 28 December 2015).
Responding to the threat of antimicrobial resistance: Australia’s first national antimicrobial resistance strategy 2015–2019, Departments of Health and Agriculture, Australian Government, June 2015. http://www.health.gov.au/internet/main/publishing.nsf/Content/1803C433C71415CACA257C8400121B1F/$File/amr-strategy-2015-2019.pdf (Accessed 28 December 2015).
Dominey-Howes, D., Michael, C. & Labbate, M. Why emergency management should be interested in the emergence of antibiotic resistance. Aust. J. Emerg. Manage 29, 11–15 (2014).
Ventola, C. L. The antibiotic resistance crisis Part 1: causes and threats. P&T 40, 277–283 (2015).
Ventola, C. L. The antibiotic resistance crisis Part 2: management strategies and new agents. P&T 40, 344–352 (2015).
Rossolini, G. M., Arena, F., Pecile, P. & Pollini, S. Update on the antibiotic resistance crisis. Curr. Opin. Pharmacol. 18, 56–60 (2014).
Michael, C. A., Dominey-Howes, D. & Labbate, M. The antimicrobial resistance crisis: causes, consequences and management. Front. Public Health 2, 145 (2014).
O'Neill, J. UK review on antimicrobial resistance. Antimicrobial resistance: tackling a crisis for the health and wealth of nations, December 2014. http://amr-review.org/Publications (Accessed 28 December 2015).
Shallcross, L. J., Howard, S. J., Fowler, T. & Davies, S. C. Tackling the threat of antimicrobial resistance: from policy to sustainable action. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20140082 (2015).
Pea, F. Editorial overview: Anti-infectives: current challenges and unmet needs in antimicrobial therapy. Curr. Opin. Pharmacol. 24, iv–vi (2015).
Woolhouse, M. E. J. & Ward, M. J. Sources of antimicrobial resistance. Science 341, 1460–1461 (2013).
Johnson, A. P. Surveillance of antibiotic resistance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20140080 (2015).
Meek, R. W., Vyas, H. & Piddock, L. J. V. Nonmedical uses of antibiotics: time to restrict their use? PLoS Biol. 13, e1002266 (2015).
Woolhouse, M., Ward, M., van Bunnik, B. & Farrar, J. Antimicrobial resistance in humans, livestock and the wider environment. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20140083 (2015).
Aarestrup, F. M. The livestock reservoir for antimicrobial resistance: a personal view on changing patterns of risks, effects of interventions and the way forward. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20140085 (2015).
Fernandes, P. The global challenge of new classes of antibacterial agents: an industry perspective. Curr. Opin. Pharmacol. 24, 7–11 (2015).
Anderson, R. M. Preface. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20140305 (2015).
Servick, K. The drug push. Science 348, 850–853 (2015).
Tommasi, R., Brown, D. G., Walkup, G. K., Manchester, J. I. & Miller, A. A. ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug Discov. 14, 529–542 (2015).
Walsh, C. T. & Wencewicz, T. A. Prospects for new antibiotics: a molecule-centered perspective. J. Antibiot. 67, 7–22 (2014).
Singh, S. B. Confronting the challenges of discovery of novel antibacterial agents. Bioorg. Med. Chem. Lett. 24, 3683–3689 (2014).
Bettiola, E. & Harbarth, S. Development of new antibiotics: taking off finally? Swiss Med. Wkly 145, w14167 (2015).
Brooks, B. D. & Brooks, A. E. Therapeutic strategies to combat antibiotic resistance. Adv. Drug Delivery Rev 78, 14–27 (2014).
O'Connell, K. M. G. et al. Combating multidrug-resistant bacteria: current strategies for the discovery of novel antibacterials. Angew. Chem. Int. Ed. Engl. 52, 10706–10733 (2013).
Shapiro, S. Speculative strategies for new antibacterials: all roads should not lead to Rome. J. Antibiot. 66, 371–386 (2013).
Smolentsev, A. I., Lavrenova, L. G., Elokhina, V. N., Nakhmanovich, A. S. & Larina, L. I. Crystal structures of pyridine-4-aldehyde thiosemicarbazone perchlorate and trifluoromethane sulfonate. J. Struct. Chem. 50, 500–504 (2009).
Grunberg, E. & Leiwant, B. Antituberculous activity in vivo of nicotinaldehyde thiosemicarbazone and its isomers. Proc. Soc. Exp. Biol. Med. 77, 47–50 (1951).
Fox, H. H. Synthetic tuberculostats. III. Isonicotinaldehyde thiosemicarbazone and some related compounds. J. Org. Chem. 17, 555–562 (1952).
Nunn, P., Porter, J. & Winstanley, P. Thiacetazone—avoid like poison or use with care? Trans. R. Soc. Trop. Med. Hyg. 87, 578–582 (1993).
Gopal, P. & Dick, T. The new tuberculosis drug Perchlozone® shows cross-resistance with thiacetazone. Int. J. Antimicrob. Agents 45, 430–433 (2015).
Blair, H. A. & Scott, L. J. Delamanid: a review of its use in patients with multidrug-resistant tuberculosis. Drugs 75, 91–100 (2015).
Gupta, R. et al. Delamanid for extensively drug-resistant tuberculosis. N. Engl. J. Med. 373, 291–292 (2015).
Sasaki, H. et al. Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles. J. Med. Chem. 49, 7854–7860 (2006).
Matsumoto, M. et al. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 3, 2131–2144 (2006).
Ashtekar, D. R. et al. In vitro and in vivo activities of the nitroimidazole CGI 17341 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 37, 183–186 (1993).
Gurumurthy, M. et al. Substrate specificity of the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis responsible for the bioreductive activation of bicyclic nitroimidazoles. FEBS J. 279, 113–125 (2012).
Butler, M. S., Hansford, K. A., Blaskovich, M. A., Halai, R. & Cooper, M. A. Glycopeptide antibiotics: back to the future. J. Antibiot. 67, 631–644 (2014).
Zhanel, G. et al. New lipoglycopeptides. Drugs 70, 859–886 (2010).
Guskey, M. T. & Tsuji, B. T. A comparative review of the lipoglycopeptides: oritavancin, dalbavancin, and telavancin. Pharmacotherapy 30, 80–94 (2010).
Anderson, V. R. & Keating, G. M. Adis drug profile—dalbavancin. Drugs 68, 639–648 (2008).
Actavis receives approval from the European commission for XYDALBA (dalbavancin) (press release 2 March 2015). http://www.allergan.com/NEWS/News/Thomson-Reuters/Actavis-Receives-Approval-from-the-European-Commis (Accessed 28 December 2015).
FDA accepts supplemental New Drug Application (sNDA) for DALVANCE (dalbavancin), 9 October 2015. http://www.allergan.com/NEWS/News/Thomson-Reuters/FDA-Accepts-Supplemental-New-Drug-Application-sNDA (Accessed 28 December 2015).
Actavis announces topline phase 3 clinical trial results for single-dose DALVANCE (dalbavancin) in the treatment of ABSSSI (press release 24 April 2015). http://www.allergan.com/news/news/thomson-reuters/actavis-announces-topline-phase-3-clinical-trial-r (Accessed 28 December 2015).
Cheng, M. et al. Anti-cooperative ligand binding and dimerisation in the glycopeptide antibiotic dalbavancin. Org. Biomol. Chem. 12, 2568–2575 (2014).
Fox, J. L. Second MRSA antibiotic reaches the market. Nat. Biotechnol. 32, 972–972 (2014).
Bouza, E. & Burillo, A. Oritavancin: a novel lipoglycopeptide active against Gram-positive pathogens including multiresistant strains. Int. J. Antimicrob. Agents 36, 401–407 (2010).
The Medicines Company receives European commission approval for three hospital acute care products: KENGREXAL (cangrelor), ORBACTIV (oritavancin) and RAPLIXA (sealant powder), 30 March 2015. http://www.themedicinescompany.com/investors/news/medicines-company-receives-european-commission-approval-three-hospital-acute-care (Accessed 28 December 2015).
Kim, S. J. et al. Oritavancin exhibits dual mode of action to inhibit cell-wall biosynthesis, in Staphylococcus aureus. J. Mol. Biol. 377, 281–293 (2008).
Patti, G. J. et al. Vancomycin and oritavancin have different modes of action in Enterococcus faecium. J. Mol. Biol. 392, 1178–1191 (2009).
Belley, A. et al. Oritavancin disrupts membrane integrity of Staphylococcus aureus and vancomycin-resistant Enterococci to effect rapid bacterial killing. Antimicrob. Agents Chemother. 54, 5369–5371 (2010).
Allen, N. E. & Nicas, T. I. Mechanism of action of oritavancin and related glycopeptide antibiotics. FEMS Microbiol. Rev. 26, 511–532 (2003).
Zhanel, G. G., Schweizer, F. & Karlowsky, J. A. Oritavancin: mechanism of action. Clin. Infect. Dis. 54, S214–S219 (2012).
Kim, S. J., Tanaka, K. S. E., Dietrich, E., Far, A. R. & Schaefer, J. Locations of the hydrophobic side chains of lipoglycopeptides bound to the peptidoglycan of Staphylococcus aureus. Biochemistry 52, 3405–3414 (2013).
Ong, V. et al. Absorption, distribution, metabolism, and excretion of the novel antibacterial prodrug tedizolid phosphate. Drug Metab. Dispos. 42, 1275–1284 (2014).
Burdette, S. D. & Trotman, R. Tedizolid: the first once-daily oxazolidinone class antibiotic. Clin. Infect. Dis. 61, 1315–1321 (2015).
Zhanel, G. et al. Tedizolid: a novel oxazolidinone with potent activity against multidrug-resistant Gram-positive pathogens. Drugs 75, 253–270 (2015).
Rodríguez-Avial, I. et al. In vitro activity of tedizolid (TR-700) against linezolid-resistant staphylococci. J. Antimicrob. Chemother. 67, 167–169 (2012).
Im, W. B. et al. Discovery of torezolid as a novel 5-hydroxymethyl-oxazolidinone antibacterial agent. Eur. J. Med. Chem. 46, 1027–1039 (2011).
Prokocimer, P., De Anda, C., Fang, E., Mehra, P. & Das, A. Tedizolid phosphate vs linezolid for treatment of acute bacterial skin and skin structure infections: the establish-1 randomized trial. JAMA 309, 559–569 (2013).
Locke, J. B., Hilgers, M. & Shaw, K. J. Novel ribosomal mutations in Staphylococcus aureus strains identified through selection with the oxazolidinones linezolid and torezolid (TR-700). Antimicrob. Agents Chemother. 53, 5265–5274 (2009).
Sucher, A. J., Chahine, E. B., Cogan, P. & Fete, M. Ceftolozane/tazobactam: a new cephalosporin and β-lactamase inhibitor combination. Ann. Pharmacother. 49, 1046–1056 (2015).
Lucasti, C. et al. Multicenter, double-blind, randomized, phase II trial to assess the safety and efficacy of ceftolozane-tazobactam plus metronidazole compared with meropenem in adult patients with complicated intra-abdominal infections. Antimicrob. Agents Chemother. 58, 5350–5357 (2014).
Wagenlehner, F. M., Umeh, O., Steenbergen, J., Yuan, G. & Darouiche, R. O. Ceftolozane-tazobactam compared with levofloxacin in the treatment of complicated urinary-tract infections, including pyelonephritis: a randomised, double-blind, phase 3 trial (ASPECT-cUTI). Lancet 385, 1949–1956 (2015).
Toda, A. et al. Synthesis and SAR of novel parenteral anti-pseudomonal cephalosporins: Discovery of FR264205. Bioorg. Med. Chem. Lett. 18, 4849–4852 (2008).
Takeda, S., Nakai, T., Wakai, Y., Ikeda, F. & Hatano, K. In vitro and in vivo activities of a new cephalosporin, FR264205, against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 51, 826–830 (2007).
Melchers, M. J. B., van Mil, A. C. H. A. M. & Mouton, J. W. In vitro activity of ceftolozane alone and in combination with tazobactam against extended-spectrum-β-lactamase-harboring Enterobacteriaceae. Antimicrob. Agents Chemother. 59, 4521–4525 (2015).
Micetich, R. G. et al. Synthesis and β-lactamase inhibitory properties of 2β-[(1,2,3-triazol-1-yl)methyl]-2α-methylpenam-3α-carboxylic acid 1,1-dioxide and related triazolyl derivatives. J. Med. Chem. 30, 1469–1474 (1987).
Gin, A. et al. Piperacillin-tazobactam: a β-lactam/β-lactamase inhibitor combination. Expert Rev. Anti Infect. Ther. 5, 365–383 (2007).
Poole, R. M. Nemonoxacin: first global approval. Drugs 74, 1445–1453 (2014).
Huang, C.-H., Lai, C.-C., Chen, Y.-H. & Hsueh, P.-R. The potential role of nemonoxacin for treatment of common infections. Expert Opin. Pharmacother. 16, 263–270 (2014).
TaiGen on the way to sell patented drug in China, Taiwan News (press release 7 August 2015). http://www.taiwannews.com.tw/etn/news_content.php?id=2783401 (Accessed 8 October 2015).
Hsu, M.-S. et al. In vitro susceptibilities of clinical isolates of ertapenem-non-susceptible Enterobacteriaceae to nemonoxacin, tigecycline, fosfomycin and other antimicrobial agents. Int. J. Antimicrob. Agents 37, 276–278 (2011).
Lauderdale, T.-L., Shiau, Y.-R., Lai, J.-F., Chen, H.-C. & King, C.-H. R. Comparative in vitro activities of nemonoxacin (TG-873870), a novel nonfluorinated quinolone, and other quinolones against clinical isolates. Antimicrob. Agents Chemother. 54, 1338–1342 (2010).
Chotikanatis, K., Kohlhoff, S. A. & Hammerschlag, M. R. In vitro activity of nemonoxacin, a novel nonfluorinated quinolone antibiotic, against Chlamydia trachomatis and Chlamydia pneumoniae. Antimicrob. Agents Chemother. 58, 1800–1801 (2014).
McKeage, K. Finafloxacin: first global approval. Drugs 75, 687–693 (2015).
MerLion’s finafloxacin shown to be more efficacious than ciprofloxacin in the treatment of complicated urinary tract infections (press release 20 September 2015). http://www.merlionpharma.com/?q=node/231 (Accessed 14 October 2015).
Dalhoff, A., Schubert, S. & Ullmann, U. Effect of pH on the in vitro activity of and propensity for emergence of resistance to fluoroquinolones, macrolides, and a ketolide. Infection 33, 36–43 (2005).
Emrich, N.-C., Heisig, A., Stubbings, W., Labischinski, H. & Heisig, P. Antibacterial activity of finafloxacin under different pH conditions against isogenic strains of Escherichia coli expressing combinations of defined mechanisms of fluoroquinolone resistance. J. Antimicrob. Chemother. 65, 2530–2533 (2010).
Stubbings, W. et al. In Vitro spectrum of activity of finafloxacin, a novel, pH-activated fluoroquinolone, under standard and acidic conditions. Antimicrob. Agents Chemother. 55, 4394–4397 (2011).
Lee, J. W. et al. High efficacy of finafloxacin on Helicobacter pylori isolates at pH 5.0 compared with those of other fluoroquinolones. Antimicrob. Agents Chemother. 59, 7629–7636 (2015).
Richards, D. M. & Brogden, R. N. Ceftazidime. A review of its antibacterial activity, pharmacokinetic properties and therapeutic use. Drugs 29, 105–161 (1985).
Bonnefoy, A. et al. In vitro activity of AVE1330A, an innovative broad-spectrum non-β-lactam β-lactamase inhibitor. J. Antimicrob. Chemother. 54, 410–417 (2004).
Stachyra, T. et al. Mechanistic studies of the inactivation of TEM-1 and P99 by NXL104, a novel non-β-lactam β-lactamase inhibitor. Antimicrob. Agents Chemother. 54, 5132–5138 (2010).
Ehmann, D. E. et al. Avibactam is a covalent, reversible, non–β-lactam β-lactamase inhibitor. Proc. Natl Acad. Sci. USA 109, 11663–11668 (2012).
Maruho Co., Ltd. Maruho receives manufacturing and marketing approval for quinolone anti-microbial topical treatment Zebiax Lotion 2% (press release 28 September 2015). https://www.maruho.co.jp/english/release/rvcck40000006x3o-att/20150928_pr_eng.pdf (Accessed 26 November 2015).
Yamakawa, T., Mitsuyama, J. & Hayashi, K. In vitro and in vivo antibacterial activity of T-3912, a novel non-fluorinated topical quinolone. J. Antimicrob. Chemother. 49, 455–465 (2002).
López, Y. et al. In vitro activity of ozenoxacin against quinolone-susceptible and quinolone-resistant Gram-positive bacteria. Antimicrob. Agents Chemother. 57, 6389–6392 (2013).
Medimetriks Pharmaceuticals, Inc. announces that the second phase 3 study for ozenoxacin has been initiated by Ferrer (press release 12 June 2014). http://www.biospace.com/News/medimetriks-pharmaceuticals-inc-announces-that-the/336637 (Accessed 5 January 2016).
Cipher Pharmaceuticals acquires Canadian commercialization rights to novel antibacterial compound Ozenoxacin (press release 7 January 2015). http://www.prnewswire.com/news-releases/cipher-pharmaceuticals-acquires-canadian-commercialization-rights-to-novel-antibacterial-compound-ozenoxacin-287771711.html (Accessed 5 January 2016).
Garde, D. Cempra tanks on 'positive' Phase III for pneumonia antibiotic, 16 October 2015. http://www.fiercebiotech.com/story/cempra-tanks-positive-phase-iii-pneumonia-antibiotic/2015-10-16 (Accessed 28 December 2015).
Rodgers, W., Frazier, A. D. & Champney, W. S. Solithromycin inhibition of protein synthesis and ribosome biogenesis in Staphylococcus aureus Streptococcus pneumoniae, and Haemophilus influenzae. Antimicrob. Agents Chemother. 57, 1632–1637 (2013).
Farrell, D. J., Castanheira, M., Sader, H. S. & Jones, R. N. The in vitro evaluation of solithromycin (CEM-101) against pathogens isolated in the United States and Europe (2009). J. Infect. 61, 476–483 (2010).
Putnam, S. D., Sader, H. S., Farrell, D. J., Biedenbach, D. J. & Castanheira, M. Antimicrobial characterisation of solithromycin (CEM-101), a novel fluoroketolide: activity against staphylococci and enterococci. Int. J. Antimicrob. Agents 37, 39–45 (2011).
Golparian, D., Fernandes, P., Ohnishi, M., Jensen, J. S. & Unemo, M. In vitro activity of the new fluoroketolide solithromycin (CEM-101) against a large collection of clinical Neisseria gonorrhoeae isolates and international reference strains, including those with high-level antimicrobial resistance: potential treatment option for gonorrhea? Antimicrob. Agents Chemother. 56, 2739–2742 (2012).
Piccinelli, G. et al. In vitro activity of solithromycin against erythromycin-resistant Streptococcus agalactiae. Antimicrob. Agents Chemother. 58, 1693–1698 (2014).
Jensen, J. S., Fernandes, P. & Unemo, M. In vitro activity of the new fluoroketolide solithromycin (CEM-101) against macrolide-resistant and -susceptible Mycoplasma genitalium strains. Antimicrob. Agents Chemother. 58, 3151–3156 (2014).
Kobayashi, Y. et al. A novel macrolide solithromycin exerts superior anti-inflammatory effect via NF-κB inhibition. J. Pharmacol. Exp. Ther. 345, 76–84 (2013).
Carroll, J. Cempra wins $58M BARDA contract for lead antibiotic, 29 May 2013. http://www.fiercebiotech.com/story/cempra-wins-58m-barda-contract-lead-antibiotic/2013-05-29 (Accessed 28 December 2015).
Sinha, G. BARDA to pick and choose next-generation antibiotics. Nat. Biotechnol. 31, 665 (2013).
Honeyman, L. et al. Structure-activity relationship of the aminomethylcyclines and the discovery of omadacycline. Antimicrob. Agents Chemother. 59, 7044–7053 (2015).
Macone, A. B. et al. In vitro and in vivo antibacterial activities of omadacycline, a novel aminomethylcycline. Antimicrob. Agents Chemother. 58, 1127–1135 (2014).
Draper, M. P. et al. Mechanism of action of the novel aminomethylcycline antibiotic omadacycline. Antimicrob. Agents Chemother. 58, 1279–1283 (2014).
Kim, O., Assefa, H. & Honeyman, L. Paratek Pharmaceuticals. Substituted tetracycline compounds. U.S. Patent 8,318,706, 27 November (2012).
Xiao, X.-Y. et al. Fluorocyclines. 1. 7-Fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline: a potent, broad spectrum antibacterial agent. J. Med. Chem. 55, 597–605 (2012).
Clark, R. B. et al. Fluorocyclines. 2. Optimization of the C-9 side-chain for antibacterial activity and oral efficacy. J. Med. Chem. 55, 606–622 (2012).
Ronn, M. et al. Process R&D of eravacycline: the first fully synthetic fuorocycline in clinical development. Org. Process Res. Dev. 17, 838–845 (2013).
Grossman, T. H. et al. Target- and resistance-based mechanistic studies with TP-434, a novel fluorocycline antibiotic. Antimicrob. Agents Chemother. 56, 2559–2564 (2012).
Grossman, T. H., O'Brien, W., Kerstein, K. O. & Sutcliffe, J. A. Eravacycline (TP-434) is active in vitro against biofilms formed by uropathogenic Escherichia coli. Antimicrob. Agents Chemother. 59, 2446–2449 (2015).
Sutcliffe, J. A., O'Brien, W., Fyfe, C. & Grossman, T. H. Antibacterial activity of eravacycline (TP-434), a novel fluorocycline, against hospital and community pathogens. Antimicrob. Agents Chemother. 57, 5548–5558 (2013).
Tetraphase announces positive top-line results from phase 3 IGNITE 1 clinical trial of Eravacycline in complicated intra-abdominal infections, 17 December 2014. http://ir.tphase.com/releasedetail.cfm?ReleaseID=888162 (Accessed 28 December 2015).
Tetraphase announces top-line results from IGNITE2 phase 3 clinical trial of Eravacycline in cUTI, 8 September 2015. http://ir.tphase.com/releasedetail.cfm?ReleaseID=930613 (Accessed 28 December 2015).
FDA grants QIDP designation to eravacycline. Tetraphase's lead antibiotic product candidate, 15 July 2013. http://www.fiercebiotech.com/press-releases/fda-grants-qidp-designation-eravacycline-tetraphases-lead-antibiotic-product-candidate (Accessed 28 December 2015).
Yin, N. et al. Structure-activity relationship studies of a series of semisynthetic lipopeptides leading to the discovery of surotomycin, a novel cyclic lipopeptide being developed for the treatment of Clostridium difficile-associated diarrhea. J. Med. Chem. 58, 5137–5142 (2015).
Knight-Connoni, V., Mascio, C., Chesnel, L. & Silverman, J. Discovery and development of surotomycin for the treatment of Clostridium difficile. J. Ind. Microbiol. Biotechnol. 43, 195–204 (2015).
Snydman, D. R., Jacobus, N. V. & McDermott, L. A. Activity of a novel cyclic lipopeptide, CB-183,315, against resistant Clostridium difficile and other Gram-positive aerobic and anaerobic intestinal pathogens. Antimicrob. Agents Chemother. 56, 3448–3452 (2012).
Citron, D. M., Tyrrell, K. L., Merriam, C. V. & Goldstein, E. J. C. In vitro activities of CB-183,315, vancomycin, and metronidazole against 556 strains of Clostridium difficile, 445 other intestinal anaerobes, and 56 Enterobacteriaceae species. Antimicrob. Agents Chemother. 56, 1613–1615 (2012).
Bouillaut, L. et al. Effects of surotomycin on Clostridium difficile viability and toxin production in vitro. Antimicrob. Agents Chemother. 59, 4199–4205 (2015).
Alam, M. Z., Wu, X., Mascio, C., Chesnel, L. & Hurdle, J. G. Mode of action and bactericidal properties of surotomycin against growing and nongrowing Clostridium difficile. Antimicrob. Agents Chemother. 59, 5165–5170 (2015).
Citron, D. M., Tyrrell, K. L., Dale, S. E., Chesnel, L. & Goldstein, E. J. C. Impact of surotomycin on the gut microbiota of healthy volunteers in a phase 1 clinical trial. Antimicrob. Agents Chemother. 60, 2069–2074 (2016).
Zhanel, G. G. et al. Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin. Expert Rev. Anti-infect. Ther 10, 459–473 (2012).
Karaiskos, I., Souli, M. & Giamarellou, H. Plazomicin: an investigational therapy for the treatment of urinary tract infections. Expert Opin. Invest. Drugs 24, 1501–1511 (2015).
Aggen, J. B. et al. Synthesis and spectrum of the neoglycoside ACHN-490. Antimicrob. Agents Chemother. 54, 4636–4642 (2010).
Weinstein, M. J. et al. Antibiotic 6640, a new Micromonospora-produced aminoglycoside antibiotic. J. Antibiot. 23, 551–554 (1970).
Reimann, H. et al. Structure of sisomicin, a novel unsaturated aminocyclitol antibiotic from Micromonospora inyoensis. J. Org. Chem. 39, 1451–1457 (1974).
Galani, I. et al. Activity of plazomicin (ACHN-490) against MDR clinical isolates of Klebsiella pneumoniae Escherichia coli, and Enterobacter spp. from Athens, Greece. J. Chemother. 24, 191–194 (2012).
Almaghrabi, R. et al. Carbapenem-resistant Klebsiella pneumoniae strains exhibit diversity in aminoglycoside-modifying enzymes, which exert differing effects on plazomicin and other agents. Antimicrob. Agents Chemother. 58, 4443–4451 (2014).
Walkty, A. et al. In vitro activity of plazomicin against 5015 Gram-negative and Gram-positive clinical isolates obtained from patients in Canadian hospitals as part of the CANWARD study, 2011-2012. Antimicrob. Agents Chemother. 58, 2554–2563 (2014).
Olsen, S. C. & Carlson, S. A. In vitro bactericidal activity of aminoglycosides, including the next-generation drug plazomicin, against Brucella spp. Int. J. Antimicrob. Agents 45, 76–78 (2015).
R-Pharm Group of Companies continues development of new generation antibiotic (press release 18 March 2015). http://r-pharm.com/en/news/article-242/ (Accessed 15 December 2015).
Stryjewski, M. E. et al. TD-1792 versus vancomycin for treatment of complicated skin and skin structure infections. Antimicrob. Agents Chemother. 56, 5476–5483 (2012).
Blais, J., Lewis, S. R., Krause, K. M. & Benton, B. M. Antistaphylococcal activity of TD-1792, a multivalent glycopeptide-cephalosporin antibiotic. Antimicrob. Agents Chemother. 56, 1584–1587 (2012).
Lee, R. E. et al. Combinatorial lead optimization of [1,2]-diamines based on ethambutol as potential antituberculosis preclinical candidates. J. Comb. Chem. 5, 172–187 (2003).
Maxwell Biotech Venture Fund’s Portfolio Company, Infectex, enrolls first multi-drug resistant tuberculosis (MDR-TB) patients in pivotal clinical trial of SQ109, licensed from Sequella (press release 19 December 2012). http://www.sequella.com/docs/Sequella_Infectex_Release_19Dec2012.pdf (Accessed 6 January 2016).
Li, W. et al. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 6413–6423 (2014).
Li, K. et al. Oxa, thia, heterocycle, and carborane analogues of SQ109: bacterial and protozoal cell growth inhibitors. ACS Infect. Dis. 1, 215–221 (2015).
Veiga-Santos, P. et al. SQ109, a new drug lead for Chagas disease. Antimicrob. Agents Chemother. 59, 1950–1961 (2015).
Locher, H. H. et al. Investigations of the mode of action and resistance development of cadazolid, a new antibiotic for treatment of Clostridium difficile infections. Antimicrob. Agents Chemother. 58, 901–908 (2014).
Rashid, M.-U., Lozano, H. M., Weintraub, A. & Nord, C. E. In vitro activity of cadazolid against Clostridium difficile strains isolated from primary and recurrent infections in Stockholm, Sweden. Anaerobe 20, 32–35 (2013).
Locher, H. H. et al. In vitro and in vivo antibacterial evaluation of cadazolid, a new antibiotic for treatment of Clostridium difficile infections. Antimicrob. Agents Chemother. 58, 892–900 (2014).
Stover, C. K. et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405, 962–966 (2000).
Singh, R. et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322, 1392–1395 (2008).
Manjunatha, U., Boshoff, H. I. & Barry, C. E. The mechanism of action of PA-824: novel insights from transcriptional profiling. Commun. Integr. Biol. 2, 215–218 (2009).
Haver, H. L. et al. Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 59, 5316–5323 (2015).
Van Bambeke, F. Delafloxacin, a non-zwitterionic fluoroquinolone in Phase III of clinical development: evaluation of its pharmacology, pharmacokinetics, pharmacodynamics and clinical efficacy. Future Microbiol. 10, 1111–1123 (2015).
Melinta Therapeutics presents complete delafloxacin results from phase 3 study in patients with acute bacterial skin and skin structure infections at ID week, 9 October 2015. http://www.melinta.com/news.php?c=39 (Accessed 28 December 2015).
Lemaire, S., Tulkens, P. M. & Van, B. F. Contrasting effects of acidic pH on the extracellular and intracellular activities of the anti-gram-positive fluoroquinolones moxifloxacin and delafloxacin against Staphylococcus aureus. Antimicrob. Agents Chemother. 55, 649–658 (2011).
Drlica, K. et al. Quinolones: action and resistance updated. Curr. Top. Med. Chem. 9, 981–998 (2009).
Araya, I., Goto, A., Minagawa, W., Funada, K. & Nagao, M. Kyorin Pharmaceutical Co., Ltd. Preparation of 7-[(3S,4S-3-[(cyclopropylamino)methyl]-4-fluoropyrrolidin-1-yl]-6-fluoro-1-(2-fluoroethyl)-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid salt crystals with high photostability and storage stability. US Patent 9,090,587, 28 July (2015).
Main R&D Activities (as of November 5 2015). http://www.kyorin-pharm.co.jp/en/business/pdf/main_rd_activities_20151105_en.pdf (Accessed 25 November 2015).
CK, R. et al. Zabofloxacin versus moxifloxacin in patients with COPD exacerbation: a multicenter, double-blind, double-dummy, randomized, controlled. Phase III, non-inferiority trial. Int. J. Chron. Obstruct. Pulmon. Dis. 10, 2265–2275 (2015).
XF-73 (Destiny Pharmaceuticals Website). http://www.destinypharma.com/xf73.shtml (Accessed 26 November 2015).
Maisch, T., Bosl, C., Szeimies, R. M., Lehn, N. & Abels, C. Photodynamic effects of novel XF porphyrin derivatives on prokaryotic and eukaryotic cells. Antimicrob. Agents Chemother. 49, 1542–1552 (2005).
Ooi, N. et al. XF-73, a novel antistaphylococcal membrane-active agent with rapid bactericidal activity. J. Antimicrob. Chemother. 64, 735–740 (2009).
Farrell, D. J., Robbins, M., Rhys-Williams, W. & Love, W. G. In vitro activity of XF-73, a novel antibacterial agent, against antibiotic-sensitive and -resistant Gram-positive and Gram-negative bacterial species. Int. J. Antimicrob. Agents 35, 531–536 (2010).
Ooi, N. et al. XF-70 and XF-73, novel antibacterial agents active against slow-growing and non-dividing cultures of Staphylococcus aureus including biofilms. J. Antimicrob. Chemother. 65, 72–78 (2010).
Farrell, D. J., Robbins, M., Rhys-Williams, W. & Love, W. G. Investigation of the potential for mutational resistance to XF-73, retapamulin, mupirocin, fusidic acid, daptomycin, and vancomycin in methicillin-resistant Staphylococcus aureus isolates during a 55-passage study. Antimicrob. Agents Chemother. 55, 1177–1181 (2011).
Gonzales, F. P., Felgenträger, A., Bäumler, W. & Maisch, T. Fungicidal photodynamic effect of a twofold positively charged porphyrin against Candida albicans planktonic cells and biofilms. Future Microbiol. 8, 785–797 (2013).
Shiau, T. P. et al. Stieglitz rearrangement of N,N-dichloro-β,β-disubstituted taurines under mild aqueous conditions. Bioorg. Med. Chem. Lett. 19, 1110–1114 (2009).
Francavilla, C. et al. Quaternary ammonium N,N-dichloroamines as topical, antimicrobial agents. Bioorg. Med. Chem. Lett. 19, 2731–2734 (2009).
Wang, L., Khosrovi, B. & Najafi, R. N-Chloro-2,2-dimethyltaurines: a new class of remarkably stable N-chlorotaurines. Tetrahedron Lett. 49, 2193–2195 (2008).
Gottardi, W., Debabov, D. & Nagl, M. N-chloramines, a promising class of well-tolerated topical anti-infectives. Antimicrob. Agents Chemother. 57, 1107–1114 (2013).
Jekle, A. et al. NVC-422 inactivates Staphylococcus aureus toxins. Antimicrob. Agents Chemother. 57, 924–929 (2013).
Novak, R. Are pleuromutilin antibiotics finally fit for human use? Ann. N. Y. Acad. Sci. 1241, 71–81 (2011).
Novak, R. & Shlaes, D. M. The pleuromutilin antibiotics: a new class for human use. Curr. Opin. Invest. Drugs 11, 182–191 (2010).
Garde, D. Nabriva pulls off a down-sized IPO, raising $92M for antibiotic R&D, 18 September 2015. http://www.fiercebiotech.com/story/nabriva-pulls-down-sized-ipo-raising-92m-antibiotic-rd/2015-09-18 (Accessed 28 December 2015).
Prince, W. T. et al. Phase II clinical study of BC-3781, a pleuromutilin antibiotic, in treatment of patients with acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother. 57, 2087–2094 (2013).
Sader, H. S. et al. Antimicrobial activity of the novel pleuromutilin antibiotic BC-3781 against organisms responsible for community-acquired respiratory tract infections (CARTIs). J. Antimicrob. Chemother. 67, 1170–1175 (2012).
Sader, H. S., Biedenbach, D. J., Paukner, S., Ivezic-Schoenfeld, Z. & Jones, R. N. Antimicrobial activity of the investigational pleuromutilin compound BC-3781 tested against Gram-positive organisms commonly associated with acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother. 56, 1619–1623 (2012).
Cassiopea product pipeline: CB-06-01. http://www.cassiopea.com/activities/product-pipeline/cb-06-01.aspx (Accessed 28 December 2015).
Ito, A. et al. In vitro antimicrobial activity of S-649266, a catechol-substituted siderophore cephalosporin, when tested against non-fermenting Gram-negative bacteria. J. Antimicrob. Chemother. 71, 670–677 (2016).
Kohira, N. et al. In vitro antimicrobial activity of siderophore cephalosporin S-649266 against Enterobacteriaceae clinical isolates including carbapenem-resistant strains. Antimicrob. Agents Chemother. 60, 729–734 (2015).
Tsuji, M. et al S-649266, a novel sderophore cephalosporin: mechanisms of enhanced activity and beta-lactamase stability. Poster 256 ID Week 9 October 2014 https://idsa.confex.com/idsa/2014/webprogram/Handout/id2651/POSTER40_256.pdf (Accessed 28 December 2015).
Shionogi and GlaxoSmithKline to collaborate on the research, development and commercialization of novel antibiotics targeting drug-resistant Gram-negative bacteria (press release 28 October 2010). http://www.shionogi.co.jp/en/company/news/2015/pmrltj0000002hjp-att/e_151109.pdf (Accessed 28 December 2015).
Shionogi development pipeline 2013. Annual Report. http://www.shionogi.co.jp/en/ir/pdf/pdf_13/all13.pdf (Accessed 28 December 2015).
GlaxoSmithKline product development pipeline 2013, February 2013. http://www.glaxosmithkline.de/docs-pdf/forschung/GSK-product-pipeline-Feb-2013.pdf (Accessed 28 December 2015).
Srinivas, N. et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 327, 1010–1013 (2010).
Demarco, S. J. et al Polyphor, Ltd. and Universität Zürich. Template-fixed peptidomimetics with antimicrobial activity. US Patent 8,685,922, 1 April (2014).
Kokryakov, V. N. et al. Protegrins: leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 327, 231–236 (1993).
Lou, K.-J. A new spin on protegrin. SciBX 3, doi:10.1038/scibx.2010.265 (2010).
Polyphor reports successful Phase I results for its Pseudomonas selective antibiotic POL7080 (press release 4 March 2013). http://www.polyphor.com/assets/files/Press_Release/POL7080_Press%20Release_.pdf (Accessed 31 May 2013).
Roche and polyphor join efforts to combat multi-drug-resistant bacterial infections. http://www.fiercebiotech.com/press-releases/roche-and-polyphor-join-efforts-combat-multi-drug-resistant-bacterial-infec (Accessed 28 December 2015).
Carroll, J. Roche abandons a marquee antibiotics collaboration with polyphor (press release 30 November 2015). http://www.fiercebiotech.com/story/roche-abandons-marquee-antibiotics-collaboration-polyphor/2015-11-30 (Accessed 28 December 2015).
Shlaes, D. IDSA update on antibiotic development—hope or stagnation? (Published 22 April 2013). http://antibiotics-theperfectstorm.blogspot.com.au/2013/04/idsa-update-on-antibiotic-development.html (Accessed 24 May 2013).
Cellceutix acquires PolyMedix assets from bankruptcy court, gains ownership of two clinical stage drugs, multiple cmpounds, andequipment assets (press release 9 September 2013). http://www.wallstreet-online.de/nachricht/6322539-cellceutix-acquires-polymedix-assets-from-bankruptcy-court-gains-ownership-of-two-clinical-stage-drugs-multiple-compounds-and-equipment-assets (Accessed 28 December 2015).
Cellceutix announces positive top-line data from phase 2b ABSSSI trial single dose brilacidin comparable to 7 days of daptomycin (press release 23 October 2014). http://cellceutix.com/cellceutix-announces-positive-top-line-data-from-phase-2b-absssi-trial-single-dose-brilacidin-comparable-to-7-days-of-daptomycin/#sthash.lmMXDdQ9.dpbs (Accessed 28 December 2015).
Cellceutix antibiotic brilacidin receives QIDP designationfrom FDA (press release 8 December 2014. http://cellceutix.com/cellceutix-antibiotic-brilacidin-receives-qidp-designation-from-fda/#sthash.0lf3MbLm.dpbs (Accessed 28 December 2015).
Enrollment begins in cellceutix phase 2 trial of brilacidin-OM to prevent oral mucositis in patients undergoing chemoradiation (press release 26 May 2015). http://cellceutix.com/enrollment-begins-in-cellceutix-phase-2-trial-of-brilacidin-om-to-prevent-oral-mucositis-in-patients-undergoing-chemoradiation/#sthash.5WntcBjI.dpbs (Accessed 28 December 2015).
Cellceutix provides brilacidin update. New class of antibiotics to enter phase 3 (press release 30 October 2015). http://cellceutix.com/cellceutix-provides-brilacidin-update-new-class-of-antibiotics-to-enter-phase-3/#sthash.47W724vM.dpbs (Accessed 28 December 2015).
Choi, S. et al. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc. Natl Acad. Sci. USA 106, 6968–6973 (2009).
Tew, G. N., Scott, R. W., Klein, M. L. & DeGrado, W. F. De novo design of antimicrobial polymers, foldamers, and small molecules: from discovery to practical applications. Acc. Chem. Res. 43, 30–39 (2010).
Degrado, W. F. et al Polymedix, Inc. Synthetic mimetics of host defense and uses thereof. U.S. Patent 8,278,309, 2 October (2012).
Study shows Cellceutix antibiotic active against drug resistant superbug Klebsiella pneumoniae (press release 14 April 2014). http://cellceutix.com/study-shows-cellceutix-antibiotic-active-against-drug-resistant-superbug-klebsiella-pneumoniae/#sthash.2AOMIr7K.dpbs (Accessed 28 December 2015).
Isaksson, J. et al. A synthetic antimicrobial peptidomimetic (LTX 109): stereochemical impact on membrane disruption. J. Med. Chem. 54, 5786–5795 (2011).
Nilsson, A. C. et al. LTX-109 is a novel agent for nasal decolonization of methicillin-resistant and -sensitive Staphylococcus aureus. Antimicrob. Agents Chemother. 59, 145–151 (2015).
Ryge, T. S., Frimodt-Moller, N. & Hansen, P. R. Antimicrobial activities of twenty lysine-peptoid hybrids against clinically relevant bacteria and fungi. Chemotherapy 54, 152–156 (2008).
Saravolatz, L. D. et al. In vitro activities of LTX-109, a synthetic antimicrobial peptide, against methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, daptomycin-nonsusceptible, and linezolid-nonsusceptible Staphylococcus aureus. Antimicrob. Agents Chemother. 56, 4478–4482 (2012).
Lytix Biopharma shelves clinical trial with LTX-109 in diabetic foot ulcers (press release 31 October 2015). http://www.lytixbiopharma.com/news/317/252/Lytix-Biopharma-shelves-clinical-trial-with-LTX-109-in-diabetic-foot-ulcers.html (Accessed 28 December 2015).
Lawrence, L., Danese, P., DeVito, J., Franceschi, F. & Sutcliffe, J. In vitro activities of the Rx-01 oxazolidinones against hospital and community pathogens. Antimicrob. Agents Chemother. 52, 1653–1662 (2008).
Zhou, J. et al. Design at the atomic level: generation of novel hybrid biaryloxazolidinones as promising new antibiotics. Bioorg. Med. Chem. Lett. 18, 6179–6183 (2008).
Zhou, J. et al. Design at the atomic level: design of biaryloxazolidinones as potent orally active antibiotics. Bioorg. Med. Chem. Lett. 18, 6175–6178 (2008).
Skripkin, E. et al. Rχ-01, a new family of oxazolidinones that overcome ribosome-based linezolid resistance. Antimicrob. Agents Chemother. 52, 3550–3557 (2008).
Malin Corporation plc 2015 Annual Report. http://www.malinplc.com/wp-content/uploads/2015/05/Malin-Annual-Report-2015.pdf (Accessed 1 June 2016).
Gordeev, M. F. & Yuan, Z. Y. Y. New potent antibacterial oxazolidinone (MRX-I) with an improved class safety profile. J. Med. Chem. 57, 4487–4497 (2014).
Huang, Y. Q. et al. Selection and characterisation of Staphylococcus aureus mutants with reduced susceptibility to the investigational oxazolidinone MRX-I. Int. J. Antimicrob. Agents 43, 418–422 (2014).
Li, C. R. et al. In vivo antibacterial activity of MRX-I, a new oxazolidinone. Antimicrob. Agents Chemother. 58, 2418–2421 (2014).
MicuRx initiates U.S. phase 2 clinical trial for novel antibiotic MRX-I (press release 2 March 2015). http://www.micurx.com/news.htm (Accessed 5 January 2016).
Johnson, P. D. et al Summit Corporation PLC. Compounds for the treatment of Clostridium difficile associated disease U.S. Patent 8,987,308, 24 March (2015).
Positive phase 1 clinical trial results on SMT 19969 reported (press release 24 April 2013). http://www.summitplc.com/media/press-releases (Accessed 5 January 2016).
Summit Therapeutics announces novel antibiotic Ridinilazole (SMT19969) achieves statistical superiority over vancomycin in CoDIFy phase 2 clinical trial for C. difficile infection (press release 23 November 2015. http://globenewswire.com/news-release/2015/11/23/789529/10157064/en/Summit-Therapeutics-Announces-Novel-Antibiotic-Ridinilazole-SMT19969-Achieves-Statistical-Superiority-Over-Vancomycin-in-CoDIFy-Phase-2-Clinical-Trial-for-C-difficile-Infection.html (Accessed 28 December 2015).
Summit Therapeutics receives FDA fast track designation for novel antibiotic SMT19969 in the treatment of C. difficile infection (press release 8 July 2015). http://globenewswire.com/news-release/2015/07/08/750560/10140996/en/Summit-Therapeutics-Receives-FDA-Fast-Track-Designation-for-Novel-Antibiotic-SMT19969-in-the-Treatment-of-C-difficile-Infection.html (Accessed 28 December 2015).
Sattar, A., Thommes, P., Payne, L., Warn, P. & Vickers, R. J. SMT19969 for Clostridium difficile infection (CDI): in vivo efficacy compared with fidaxomicin and vancomycin in the hamster model of CDI. J. Antimicrob. Chemother. 70, 1757–1762 (2015).
So, W., Crandon, J. L. & Nicolau, D. P. Pharmacodynamic profile of GSK2140944 against methicillin-resistant Staphylococcus aureus in a murine lung infection model. Antimicrob. Agents Chemother. 59, 4956–4961 (2015).
Ross, J. E., Scangarella-Oman, N. E., Flamm, R. K. & Jones, R. N. Determination of disk diffusion and MIC quality control guidelines for GSK2140944, a novel bacterial type II topoisomerase inhibitor antimicrobial agent. J. Clin. Microbiol. 52, 2629–2632 (2014).
AstraZeneca’s novel antibiotic candidate AZD0914 given Fast Track status by US FDA (press release 3 June 2014). https://www.astrazeneca.com/our-company/media-centre/press-releases/2014/astrazeneca-novel-antibiotic-azd0914-us-fda-fast-track-03062014.html (Accessed 28 December 2015).
AstraZeneca to create stand-alone company for small molecule early-stage antibiotic R&D (press release 26 February 2015). https://www.astrazeneca.com/our-company/media-centre/press-releases/2015/astrazeneca-stand-alone-company-small-molecule-early-stage-antibiotic-research-development-26022015.html (Accessed 28 December 2015).
Kern, G. et al. Inhibition of Neisseria gonorrhoeae type II topoisomerases by the novel spiropyrimidinetrione AZD0914. J. Biol. Chem. 290, 20984–20994 (2015).
Jacobsson, S. et al. High in vitro activity of the novel spiropyrimidinetrione AZD0914, a DNA gyrase inhibitor, against multidrug-resistant Neisseria gonorrhoeae isolates suggests a new effective option for oral treatment of gonorrhea. Antimicrob. Agents Chemother. 58, 5585–5588 (2014).
Parsons, J. B. et al. Perturbation of Staphylococcus aureus gene expression by the enoyl-acyl carrier protein reductase inhibitor AFN-1252. Antimicrob. Agents Chemother. 57, 2182–2190 (2013).
Kaplan, N. et al. Mode of action, in vtro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob. Agents Chemother. 56, 5865–5874 (2012).
Lu, H. & Tonge, P. J. Inhibitors of FabI, an enzyme drug target in the bacterial fatty acid biosynthesis pathway. Acc. Chem. Res. 41, 11–20 (2008).
Gerusz, V. in Annual Reports In Medicinal Chemistry Vol. 45. (ed. John, E. M. 295–311 (Academic Press, New York, (2010).
Payne, D. J. et al. Discovery of a novel and potent class of FabI-directed antibacterial agents. Antimicrob. Agents Chemother. 46, 3118–3124 (2002).
Payne, D. J., Gwynn, M. N., Holmes, D. J. & Pompliano, D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6, 29–40 (2007).
Karlowsky, J. A. et al. In vitro activity of API-1252, a novel FabI inhibitor, against clinical isolates of Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob. Agents Chemother. 51, 1580–1581 (2007).
Karlowsky, J. A., Kaplan, N., Hafkin, B., Hoban, D. J. & Zhanel, G. G. AFN-1252, a FabI inhibitor, demonstrates a Staphylococcus-specific spectrum of activity. Antimicrob. Agents Chemother. 53, 3544–3548 (2009).
Flamm, R. K., Rhomberg, P. R., Kaplan, N., Jones, R. N. & Farrell, D. J. Activity of Debio1452, a FabI inhibitor with potent activity against Staphylococcus aureus and coagulase-negative Staphylococcus spp., including multidrug-resistant strains. Antimicrob. Agents Chemother. 59, 2583–2587 (2015).
Tsuji, B. T., Harigaya, Y., Lesse, A. J., Forrest, A. & Ngo, D. Activity of AFN-1252, a novel FabI inhibitor, against Staphylococcus aureus in an in vitro pharmacodynamic model simulating human pharmacokinetics. J. Chemother. 25, 32–35 (2013).
Rao, K. N. et al. AFN-1252 is a potent inhibitor of enoyl-ACP reductase from Burkholderia pseudomallei-Crystal structure, mode of action, and biological activity. Protein Sci. 24, 832–840 (2015).
Kaplan, N. et al. In vitro activity (MICs and rate of kill) of AFN-1252, a novel FabI inhibitor, in the presence of serum and in combination with other antibiotics. J. Chemother. 25, 18–25 (2013).
Hafkin, B., Kaplan, N. & Hunt, T. L. Safety, tolerability and pharmacokinetics of AFN-1252 administered as immediate release tablets in healthy subjects. Future Microbiol. 10, 1805–1813 (2015).
Banevicius, M. A., Kaplan, N., Hafkin, B. & Nicolau, D. P. Pharmacokinetics, pharmacodynamics and efficacy of novel FabI inhibitor AFN-1252 against MSSA and MRSA in the murine thigh infection model. J. Chemother. 25, 26–31 (2013).
Debiopharm Group to acquire Affinium’s antibiotic clinical assets and platform to identify and develop targeted antibiotics (press release 11 February 2014). https://www.debiopharm.com/medias/press-release/item/3393-debiopharm-group-to-acquire-affinium-s-antibiotic-clinical-assets-and-platform-to-identify-and-develop-targeted-antibiotics.html (Accessed 28 December 2015).
Debiopharm Group: pipeline https://www.debiopharm.com/our-business/pipeline.html (Accessed 28 December 2015).
Debiopharm Group and Nobelex Biotech start two collaborations on development of new antibiotics against N. gonorrhoeae and enteric species (press release 26 June 2014) https://www.debiopharm.com/our-business/pipeline/item/3463-debiopharm-group-and-nobelex-biotech-start-two-collaborations-on-development-of-new-antibiotics-against-n-gonorrhoeae-and-enteric-species.html (Accessed 28 December 2015).
US FDA grants breakthrough (QIDP) drug discovery status to the New Antibiotic of Wockhardt (press release 8 December 2015). http://www.wockhardt.com/pdfs/US-FDA-grants-breakthrough-drug-discovery-2015.pdf (Accessed 28 December 2015).
U.S. FDA grants QIDP status for Wockhardt anti-infective drug discovery program (press release 31 August 2014). http://www.prnewswire.com/news-releases/us-fda-grants-qidp-status-for-wockhardt-anti-infective-drug-discovery-program-273411211.html (Accessed 26 December 2015).
Jacobs, M. R. & Appelbaum, P. C. Nadifloxacin: a quinolone for topical treatment of skin infections and potential for systemic use of its active isomer, WCK 771. Expert Opin. Pharmacother. 7, 1957–1966 (2006).
de Souza, N. J. et al. A chiral benzoquinolizine-2-carboxylic acid arginine salt active against vancomycin-resistant Staphylococcus aureus. J. Med. Chem. 48, 5232–5242 (2005).
Al-Lahham, A., De Souza, N. J., Patel, M. & René Reinert, R. Activity of the new quinolones WCK 771, WCK 1152 and WCK 1153 against clinical isolates of Streptococcus pneumoniae and Streptococcus pyogenes. J. Antimicrob. Chemother. 56, 1130–1133 (2005).
Bhagwat, S. S., McGhee, P., Kosowska-Shick, K., Patel, M. V. & Appelbaum, P. C. In vtro activity of the quinolone WCK 771 against recent U.S. hospital and community-acquired Staphylococcus aureus pathogens with various resistance types. Antimicrob. Agents Chemother. 53, 811–813 (2009).
Page, M. G. P., Dantier, C. & Desarbre, E. In vitro properties of BAL30072, a novel siderophore sulfactam with activity against multiresistant Gram-negative bacilli. Antimicrob. Agents Chemother. 54, 2291–2302 (2010).
Mushtaq, S., Warner, M. & Livermore, D. Activity of the siderophore monobactam BAL30072 against multiresistant non-fermenters. J. Antimicrob. Chemother. 65, 266–270 (2010).
Basilea initiates phase I clinical program of its novel antibiotic BAL30072 (press release 23 November 2010). http://www.basilea.com/News-and-Media/Basilea-initiates-phase-I-clinical-program-of-its-novel-antibiotic-BAL30072/381 (Accessed 5 January 2016).
BARDA supports new broad-spectrum antibiotic. Drug could be first against two bioterrorism threats as well as common drug-resistant infections (press release 25 June 2013). http://www.phe.gov/Preparedness/news/Pages/broad-spectrum-June2013.aspx (Accessed 28 December 2015).
Basilea awarded contract by BARDA of up to USD 89 million for the development of its novel antibiotic BAL30072 (press release 13 June 2013). http://www.marketwired.com/press-release/basilea-awarded-contract-barda-up-usd-89-million-development-its-novel-antibiotic-bal30072-pinksheets-bpmuf-1805200.htm (Accessed 28 December 2015).
Basilea initiates phase 1 combination study with its Gram-negative antibiotic BAL30072 and meropenem (press release 12 June 2014). http://globenewswire.com/news-release/2014/06/12/643536/10085495/en/Basilea-initiates-phase-1-combination-study-with-its-Gram-negative-antibiotic-BAL30072-and-meropenem.html (Accessed 28 December 2015).
Basilea announces development of inhaled dosage form of its Gram-negative antibiotic BAL30072 as part of European iABC program (press release 7 September 2015). http://www.basilea.com/News-and-Media/Basilea-announces-development-of-inhaled-dosage-form-of-its-Gram-negative-antibiotic-BAL30072-as-part-of-European-iABC-program/eb712a99-4c9f-5acf-07f7-c8c30882a267 (Accessed 28 December 2015).
Basilea Pharmaceutica: Portfolio: BAL30072. http://www.basilea.com/Portfolio/BAL30072/ (Accessed 28 December 2015).
van Delden, C., Page, M. G. P. & Köhler, T. Involvement of Fe uptake systems and AmpC β-lactamase in susceptibility to the siderophore monosulfactam BAL30072 in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 57, 2095–2102 (2013).
Hofer, B. et al. Combined effects of the siderophore monosulfactam BAL30072 and carbapenems on multidrug-resistant Gram-negative bacilli. J. Antimicrob. Chemother. 68, 1120–1129 (2013).
Higgins, P. G., Stefanik, D., Page, M. G. P., Hackel, M. & Seifert, H. In vitro activity of the siderophore monosulfactam BAL30072 against meropenem-non-susceptible Acinetobacter baumannii. J. Antimicrob. Chemother. 67, 1167–1169 (2012).
Mima, T. et al. In vitro activity of BAL30072 against Burkholderia pseudomallei. Int. J. Antimicrob. Agents 38, 157–159 (2011).
Landman, D. et al. In vitro activity of the siderophore monosulfactam BAL30072 against contemporary Gram-negative pathogens from New York City, including multidrug-resistant isolates. Int. J. Antimicrob. Agents 43, 527–532 (2014).
Jeong, J.-W. et al. In vtro and in vivo activities of LCB01-0371, a new oxazolidinone. Antimicrob. Agents Chemother. 54, 5359–5362 (2010).
Sader, H. S., Rhomberg, P. R., Farrell, D. J., Flamm, R. K. & Jones, R. N. in 54th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) 2014, Poster F-970 (Washington, DC, USA).
Theravance Biopharma Inc. FORM 10-K United States securities and exchange commission annual report. 31 December 2014. http://investor.theravance.com/secfiling.cfm?filingID=1047469-15-2175&CIK=1583107 (Accessed 28 December 2015).
Anthony, N. G. et al. Antimicrobial lexitropsins containing amide, amidine, and alkene linking groups. J. Med. Chem. 50, 6116–6125 (2007).
MGB Biopharma: bringing true novelty to the anti-infectives space. New class of antibacterials based on a completely new mechanism of action. http://www.mgb-biopharma.com/wp-content/uploads/2013/10/MGB-Biopharma-Bio-Europe-Presentation-6th-Nov-2013.pdf (Accessed 28 December 2015).
MGB biopharma: programmes overview. http://www.mgb-biopharma.com/programs-overview-2 (Accessed 28 December 2015).
Critchley, I. A. et al. Spectrum of activity and mode of action of REP3123, a new antibiotic to treat Clostridium difficile infections. J. Antimicrob. Chemother. 63, 954–963 (2009).
NIAID launches CRS3123 Phase I trial to treat C. difficile infection (press release 10 July 2014). http://www.news-medical.net/news/20140710/NIAID-launches-CRS3123-Phase-I-trial-to-treat-C-difficile-infection.aspx (Accessed 28 December 2015).
Upton, A. M. et al. In vitro and in vivo activities of the nitroimidazole TBA-354 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 59, 136–144 (2015).
Denny, W. A. TBA-354: A new drug for the treatment of persistent tuberculosis. Chem. N.Z. 18–22 (2015). http://nzic.org.nz/CiNZ/articles/2015/CiNZ%20Jan%202015%20Denny.pdf.
Tasneen, R. et al. Contribution of the nitroimidazoles PA-824 and TBA-354 to the activity of novel regimens in murine models of tuberculosis. Antimicrob. Agents Chemother. 59, 129–135 (2015).
TB Alliance advances next-generation TB drug candidate into clinical testing. TBA-354 is the first potential tuberculosis drug to advance to Phase 1 trial in six years (press release 17 February 2015). http://www.tballiance.org/newscenter/view-brief.php?id=1118 (Accessed 28 December 2015).
Pethe, K. et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 19, 1157–1160 (2013).
Kang, S. et al. Lead optimization of a novel series of imidazo[1,2-a]pyridine amides leading to a clinical candidate (Q203) as a multi- and extensively-drug-resistant anti-tuberculosis agent. J. Med. Chem. 57, 5293–5305 (2014).
Brown, A. G. et al. Naturally-occurring β-lactamase inhibitors with antibacterial activity. J. Antibiot. 29, 668–669 (1976).
Howarth, T. T., Brown, A. G. & King, T. J. Clavulanic acid, a novel β-lactam isolated from Streptomyces clavuligerus; X-ray crystal structure analysis. J. Chem. Soc. Chem. Commun. 266–267 (1976).
Reading, C. & Cole, M. Clavulanic acid: a beta-lactamase-inhibiting beta-lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 11, 852–857 (1977).
Shlaes, D. M. New β-lactam-β-lactamase inhibitor combinations in clinical development. Ann. N. Y. Acad. Sci. 1277, 105–114 (2013).
Coleman, K. Diazabicyclooctanes (DBOs): a potent new class of non-β-lactam β-lactamase inhibitors. Curr. Opin. Microbiol. 14, 550–555 (2011).
Qin, W., Panunzio, M. & Biondi, S. β-Lactam antibiotics renaissance. Antibiotics 3, 193–215 (2014).
Livermore, D. M. & Mushtaq, S. Activity of biapenem (RPX2003) combined with the boronate β-lactamase inhibitor RPX7009 against carbapenem-resistant Enterobacteriaceae. J. Antimicrob. Chemother. 68, 1825–1831 (2013).
The Medicines Company acquires Rempex Pharmaceuticals. Novel portfolio of gram-negative antibiotics in development added to The Medicines Company's infectious disease hospital care franchise (press release 4 December 2013). http://ir.themedicinescompany.com/mobile.view?c=122204&v=203&d=1&id=1881793 (Accessed 28 December 2015).
Mangion, I. K., Ruck, R. T., Rivera, N., Huffman, M. A. & Shevlin, M. A Concise Synthesis of a β-Lactamase Inhibitor. Org. Lett. 13, 5480–5483 (2011).
Rodloff, A. C., Goldstein, E. J. C. & Torres, A. Two decades of imipenem therapy. J. Antimicrob. Chemother. 58, 916–929 (2006).
Crandon, J. L. & Nicolau, D. P. Human simulated studies of aztreonam and aztreonam-avibactam to evaluate activity against challenging Gram-negative organisms, including metallo-β-lactamase producers. Antimicrob. Agents Chemother. 57, 3299–3306 (2013).
Perros, M. AstraZeneca LabTalk: IMI launches new “New Drugs 4 Bad Bugs” projects (9 July 2013). http://www.labtalk.astrazeneca.com/collaboration/imi-launches-new-new-drugs-4-bad-bugs-projects (Accessed 28 December 2015).
Morinaka, A. et al. OP0595, a new diazabicyclooctane: mode of action as a serine β-lactamase inhibitor, antibiotic and β-lactam 'enhancer'. J. Antimicrob. Chemother. 70, 2779–2786 (2015).
Garde, D. Roche inks a $750M antibiotics deal as it re-embraces the field (13 January 2015). http://www.fiercebiotech.com/story/roche-inks-750m-antibiotics-deal-it-re-embraces-field/2015-01-13 (Accessed 28 December 2015).
Roche, Meiji and Fedora join forces to tackle increasing bacterial resistance to antibiotics: Roche licenses investigational beta-lactamase inhibitor OP0595, OP0595 targets beta-lactamase enzymes in combination with new or existing beta-lactam antibiotics to enhance their effectiveness in difficult-to-treat bacterial infections (press release 13 January 2015). http://www.roche.com/media/store/releases/med-cor-2015-01-13.htm (Accessed 28 December 2015).
Forbion Capital Partners co-leads 15 million series a financing for Allecra Therapeutics (FierceBiotech, 18 April 2013). http://www.fiercebiotech.com/press-releases/forbion-capital-partners-co-leads-15-million-series-financing-allecra-thera (Accessed 28 December 2015).
Lamonica, A., Forzatti, M. & Biondi, S. (Allecera Therapeutics SAS). Crystalline β-lactamase inhibitor. WO2015067787, 14 May (2015).
Homery, M.-C., Denot, C., Pypstra, R., Benedict, N. & Patat, A. Safety, tolerability and pharmacokinetics of AAI101, an extended-spectrum β-lactamase inhibitor, in healthy adult males. 54th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) 2004, Poster A-1339 (Washington DC, USA).
Crandon, J. L. & Nicolau, D. P. In vivo activities of simulated human doses of cefepime and cefepime-AAI101 against multidrug-resistant Gram-negative Enterobacteriaceae. Antimicrob. Agents Chemother. 59, 2688–2694 (2015).
Crandon, J. L. & Nicolau, D. P. In vitro activity of cefepime/AAI101 and comparators against cefepime non-susceptible Enterobacteriaceae. Pathogens 4, 620–625 (2015).
15th annual biotech in Europe forum, Allecra Therapeutics: product pipeline. http://www.sandtiger.co.uk/15BEF-Guide/files/assets/basic-html/index.html#57 (Accessed 28 December 2015).
New antibiotics to address anti-microbial resistance: WCK 5107 in phase 1 from Wockhardt (Pharmaceutical Intelligence, 23 November 2015). http://pharmaceuticalintelligence.com/2015/11/23/new-antibiotics-to-address-anti-microbial-resistance (Accessed 28 December 2015).
Jarvis, L. Cubist’s former research team joins Boston’s vibrant biotech ecosystem. Chem. Eng. News 90, 20–21 (2015).
BEAM to fight antimicrobial resistance. Nat. Biotechnol. 33, 889–889 (2015).
Czaplewski, L. et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect. Dis. 16, 239–251 (2016).
Brown, D. Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void? Nat. Rev. Drug Discov. 14, 821–832 (2015).
Lok, C. Mining the microbial dark matter. Nature 522, 270–273 (2015).
Owen, J. G. et al. Mapping gene clusters within arrayed metagenomic libraries to expand the structural diversity of biomedically relevant natural products. Proc. Natl Acad. Sci. USA 110, 11797–11802 (2013).
Ling, L. L. et al. A new antibiotic kills pathogens without detectable resistance. Nature 520, 388 (2015).
Fox, J. L. Fraunhofer to mine Sanofi microbial collection. Nat. Biotechnol. 32, 305–305 (2014).
Cooper, M. A. A community-based approach to new antibiotic discovery. Nat. Rev. Drug Discov. 14, 587–588 (2015).
Blaskovich, M. A. T., Zuegg, J., Elliott, A. G. & Cooper, M. A. Helping chemists discover new antibiotics. ACS Infect. Dis. 1, 285–287 (2015).
Acknowledgements
MSB and MATB are supported by the Wellcome Trust Strategic Award Scheme 066648 (the Community for Open Antimicrobial Drug Discovery), whereas MAC is an NHMRC Principal Research Fellow (APP1059354).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Butler, M., Blaskovich, M. & Cooper, M. Antibiotics in the clinical pipeline at the end of 2015. J Antibiot 70, 3–24 (2017). https://doi.org/10.1038/ja.2016.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2016.72
This article is cited by
-
Antibiotics in the clinical pipeline as of December 2022
The Journal of Antibiotics (2023)
-
Comparative genomics with evolutionary lineage in Streptomyces bacteria reveals high biosynthetic potentials
World Journal of Microbiology and Biotechnology (2023)
-
Uncovering the biodiversity and biosynthetic potentials of rare actinomycetes
Future Journal of Pharmaceutical Sciences (2022)
-
Design, characterization and structure–function analysis of novel antimicrobial peptides based on the N-terminal CATH-2 fragment
Scientific Reports (2022)
-
Bacteriophage therapy as an alternative technique for treatment of multidrug-resistant bacteria causing diabetic foot infection
International Microbiology (2022)
