
Abstract
A novel series of conjugates of the antifungal antibiotic amphotericin B (AmB) with benzoxaboroles was synthesized. Antifungal activity of new compounds was tested on yeastβ Candida albicans and Cryptococcus humicolus and filamentous fungi Aspergillus niger and Fusarium oxysporum using the broth microdilution method. The potency of di-modified derivatives against the tested strains was similar to that of the parent AmB. New derivatives demonstrated differential toxicity against human cells (colon epithelium or red blood cells). The di-modified conjugate 2-(N,N-dimethylamino)ethylamide of 3′-N-[3-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-7-yl)propanoyl] AmB (9) showed the best combination of a high antifungal activity with a low cytotoxic and hemolytic potency.
Similar content being viewed by others

Introduction
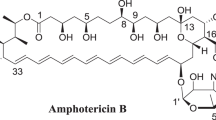
The polyene macrolide antibiotic amphotericin B (AmB) is the treatment of choice for life-threatening systemic fungal infections such as candidosis and aspergillosis. The mechanism of action of polyene macrolides is associated with their ability to interact with sterol-containing cytoplasmic membranes of pathogens, resulting in fungal cell death.1 Unfortunately, AmB therapy is limited by its severe side effects and low water solubility.
The necessity of the development of new antifungal agents for the treatment of invasive mycoses is associated with the need of drugs with less acute side effects. During the last 15 years, a variety of previously unknown semi-synthetic derivatives have been synthesized based of natural polyenes.2 The studies of the influence of structure of polyene antibiotics on their antifungal activity in vitro and in vivo demonstrated that chemical modifications of AmB as well as of the genetically engineered polyenes (at C16 carboxyl group and/or at the amino group of mycosamine moiety) may result in novel derivatives with a favorable toxicity and therapeutic efficacy.3 At the same time the differences in the structures of the polyol regions of AmB (C8, C9) or 28, 29-di-dehydro nystatin A1 (S44HP) (C7,C10) had no drastic effect on the in vitro antifungal activity.4 However, none of the newly developed polyene compounds has yet been introduced into the clinic.
Boron-containing molecules serve as drug candidates for the treatment of a variety of diseases.5, 6, 7 One of these drugs, 5-fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole developed by Anacor Pharmaceuticals (AN2690), is the first well-studied benzoxaborole antifungal agent with a broad spectrum of activity including C. neoformans and A. niger. AN2690 has received FDA approval for the treatment of onychomycosis in 2014.5 Benzoxaboroles block protein synthesis by targeting fungal leucyl-tRNA synthetase.8 The boron atom of the benzoxaborole forms a covalent tetrahedral adduct with cis-2′,3′-hydroxy groups of 3′-terminal adenosine in the editing pocket of tRNA, thus trapping the tRNA and abrogating protein synthesis.9
Moreover, the benzoxaboroles were successfully used to obtain new types of hybrid molecules, such as benzoxaborole-glycopeptides conjugates. These compounds have shown a noticeably higher activity than the starting glycopeptides against gram-positive bacteria including the resistant isolates, Staphylococci GISA and Enterococci GRE.10 A novel group of benzoxaborole–chalcone hybrids potent against parasites Trypanosoma brucei has been described.11
This study presents the synthesis and properties of a new type of hybrid antibiotics based on AmB and benzoxaboroles. Our results provide evidence that conjugation of benzoxaborole moiety to AmB can yield the compounds in which a good antifungal potency is associated with an acceptable toxicity against human cells.
Results and Discussion
Chemistry
Depending on the functional group in benzoxaborole used for the conjugation with AmB, four types of hybrid molecules were obtained: C16-amide, 3′-N-acyl, 3′-N-alkyl, 3′-N,N-bis-alkyl- and 3′-N-sulfo derivatives (Schemes 1 and 2).
The reaction of AmB (1) with (S)-3-(aminomethyl)benzo[с][1,2]oxaborol-1(3Н)-ole in the presence of PyBOP resulted in the formation of amide 2 (Scheme 1).
N-Hydroxysuccinimide esters (-OSu) of 3-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-7-yl)propionic acid, 1-hydroxy-1,3- dihydrobenzo[c][1,2]oxaborol-6-carboxylic acid or 1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-5-carboxylic acid were obtained in situ for the synthesis of 3′-N-acyl derivatives with benzoxaboroles bearing the carboxyl group in the aromatic ring. The OSu-esters were synthesized from the corresponding benzoxaboroles and N-hydroxysuccinimide in the presence of N,N'-dicyclohexylcarbodiimide (DCC). The interaction of AmB (1) with OSu-esters resulted in the formation of 3′-N-acyl derivatives 3–5 (Scheme 2).
The interaction of AmB (1) with 1-hydroxy-1,3-dihydrobenzo[с][1,2]oxaborol-6-sulfonyl chloride in the pyridine–dimethylformamide (DMF) mixture in the presence of Et3N resulted in the formation of sulfamide 6 (Scheme 2). Reductive alkylation of AmB (1) with (R)-2-(1-hydroxy-1,3-dihydrobenzo[с][1,2]oxaborol-3-yl)acetaldehyde in the presence of NaBH3CN gave a mixture of mono- and di-alkyl derivatives 7 and 8, respectively, which were separated by column chromatography on silica gel (Scheme 2).
To improve the water solubility of 3′-N-substituted derivatives of AmB, we converted compounds 3, 6 and 7 into the corresponding C16-2-(N,N-dimethylamino)ethylamides (DMAE-amides). Indeed, C-16-DMAE-amides of several polyene macrolide antibiotics of the AmB group are suitable for the preparation of the water-soluble forms.3 The C16-DMAE-amides of 3′-N-[3-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-7-yl)propanoyl] AmB (9) and 3′-N-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-6-yl)sulfonyl AmB (10) were obtained by amidation of 3 or 6, respectively, with N1,N1-dimethylethane-1,2-diamine by the standard procedure in the presence of PyBOP (Scheme 3).3
An attempt to prepare C16-DMAE-amide of 3′-N-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborole-6-methyl)-AmB (12) starting from 7 and N1,N1-dimethylethane-1,2-diamine by the same procedure failed. The target compound 12 was synthesized using an alternative method starting from C16-DMAE-amide of AmB (11) obtained by the treatment of AmB with N1,N1-dimethylethane-1,2-diamine in the presence of PyBOP. The amide 11 was converted into the derivative 12 by the reductive alkylation reaction using 1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborole-6-carbaldehyde in the presence of NaBH3CN (Scheme 4).
Purification of new compounds 2–12 was performed by column chromatography on silica gel. Yields of target compounds were low (10–25%), especially in the case of amidation reaction (compound 2, yield 10%). This can be explained by the possible formation of rather strong complexes of benzoxaboroles with the polyhydroxylated system of AmB. Yields of amidation reactions of 3 and 6, resulting in the formation of 9 and 10, respectively, (Scheme 3) were significantly higher (50%). Low yields of target compounds also can be explained by the partial destruction and irreversible sorption of AmB derivatives on silica gel during column chromatography. The purity of new compounds was determined by HPLC. The structures were confirmed by IR-, UV-spectroscopy and MS methods including the MS/MS analysis and NMR spectroscopy (Tables 1 and 2).
The UV spectra of the conjugates of AmB with benzoxaboroles 2–12 contained maximums (λmax) at 347, 365, 385 and 408 nm typical for the AmB heptaene system. The IR spectra of 2–12 contained all characteristic absorption bands, including the one at 1715–1733 cm−1(strong) corresponding to the macrolactone group. The IR spectra of amides 2, 9–12 and 3′-N-acyl derivatives 3–5 contained the absorption band at 1632–1651 cm−1 (strong) characteristic for the –CONH- group. The IR spectra of sulfamide derivatives 6 and 10 contained two absorption bands at 1161 and 1367 cm−1 (both strong) characteristic for the –SO2 group.
The negatively charged molecular peak [M–H]− was observed in the mass spectra of 3–6 and 8, which contain non-modified C-16 carboxylic group. The positively charged molecular peak [M+Н]+ was detected in the mass spectra of 2 and 9–12, which have free amino- or N-alkyl-amino group. Both negatively and positively charged peaks were observed in the mass spectrum of the mono-alkyl derivative 7.
The MS/MS fragmentation spectra also confirmed the structures of 2–12 (Table 1). Depending on the moiety at the C16-carboxylic or 3′-NH2 group, two main ways of fragmentation were observed in the MS/MS spectra.
The collision-induced dissociation of 2–12 at 40–60 eV resulted in the occurrence of the peaks with m/z that corresponded to the consecutive degradation of molecules with splitting of one and more water molecules (Table 1, Figures 1, 2, 3). In compounds 3–8 with free carboxylic group, dehydration was accompanied by decarboxylation of the starting molecular ion.

CID induced split-off of 3′-N-acyl-mycosamine moiety in 3 and the fragment of MS/MS spectrum containing the corresponding peak with m/z 332.13. CID, collision-induced dissociation.

CID induced split-off of C16-DMAE-amide aglycon fragment in 9 and the fragment of MS/MS spectrum containing the corresponding peak with m/z 813.49. CID, collision-induced dissociation; DMAE-amide, 2-(N,N-dimethylamino)ethylamide.

CID induced split-off of the C16-DMAE-amide aglycon fragment in 12 and the fragment of MS/MS spectrum containing the corresponding peak with m/z 813.49. CID, collision-induced dissociation; DMAE-amide, 2-(N,N-dimethylamino)ethylamide.
The collision-induced dissociation of 2, 3, 4, 9–12 at 30–60 eV resulted in the degradation of glycoside bond and appearance of peaks, which corresponded to the aglycon or sugar moieties in MS/MS spectra (Table 1). Typical examples of degradation (3, 9 and 12) are presented in Figures 1, 2, 3. The peak with m/z 332.13, which corresponded to the negatively charged 3′-N-acyl-mycosamine fragment was observed in the MS/MS spectra of 3, 4, confirming the attachment of benzoxaborole residue to the 3′-N-position of mycosamine (Table 1, Figure 1).
The collision-induced dissociation of C16-carboxamides 2, 9–12 resulted in β-elimination of the mycosamine moiety containing the benzoxaborole residue, which led to the positively charged peak m/z 888.48 (for compound 2) or 831.49 (for 9–12) corresponding to the dehydrated aglycon moiety (Table 1, Figures 2 and 3 for 9, 12).
The signals with m/z corresponding to the aglycon or sugar fragments were not found in the MS/MS spectra of 3′-N-substituted derivatives 5–8 (Table 2), probably due to a more stable glycoside bond.
The structure of 2–12 was confirmed by NMR spectroscopy. Introduction of benzoxaborole fragments into AmB was accompanied by the appearance of signals in the downfield (7.0–8.0 p.p.m.) region of 1H NMR spectra. It should be noted that the characteristic signal of the hydroxyl group attached to the boron atom was observed in the range 8.2 and 9.3 p.p.m. (Table 2, Supplementary Information File). For substances that contain free COOH group in the position C16 of AmB molecule, a broad signal of acidic proton was observed at 12–13 p.p.m. (Supplementary Information File).
However, taking into account the complexity of compounds, much more informative 2D NMR techniques were used to confirm their structures. At the first step, the complete assignment of the 1H and 13C resonances was obtained for 1a (3′-N-Fmoc-AmB; Scheme 2). Signal assignment was based on the analysis of cross peak correlations in DQF-COSY, TOCSY, ROESY and 13C-1H HSQC 2D NMR experiments and comparison with published partial signal assignments of AmB.12, 13 The assignment strategy was based on the direct comparison of signals in the 13C-1H HSQC spectra with analogous signals in the spectrum of 1a (Supplementary Figures S1 and S2). Signal assignment included also the analysis of DQF-COSY spectra and, in several cases, 2D ROESY spectra (Supplementary Information File).
Table 2 contains chemical shift data on AmB derivatives 1H and 13C signals, including the previously unassigned ones for 3′-N-Fmoc-AmB (1a).
Biological testing
The antifungal activity of 2–12 was tested against the yeast strains Candida albicans ATCC 14053 and Cryptococcus humicolus ATCC 9949, and against the fungi Aspergillus niger ATCC 16404 and Fusarium oxysporum VKM F-140 (=CMI, IMI 90473) using the broth microdilution method (Table 3) as described in CLSI/NCCLS documents14, 15, 16, 17 and our studies.4, 18
The majority of new derivatives was less potent than AmB (1). The antifungal activity of C16-amide of AmB with (S)-3-(aminomethyl)benzo[с][1,2]oxaborol-1(3Н)-ole (2) was lower (MICs~4–16 μg ml−1) than that of AmB (MICs~0.25–2 μg ml−1) against all the tested strains. The activities of 3′-N-acyl derivatives 3–5 and 3′-N-sulfamide 6 were slightly better (MICs~2–16 μg ml−1) but still smaller than AmB (1).
In contrast, 3′-N-mono-alkyl derivative 7 showed a significant activity against C. humicolus and A. niger (MICs=0.25 and 0.5 μg ml−1) similar to AmB. Compound 7 was slightly less active than AmB against C. albicans and F. oxysporum, demonstrating MICs 1 μg ml−1 and 4 μg ml−1, respectively, whereas for AmB, these values were 0.5 μg ml−1 and 2 μg ml−1, respectively. The 3′-N,N-di-alkyl derivative 8 was much less potent than 3′-N-mono-alkyl derivative 7.
The antifungal activity of 3 and 6 was improved by amidation of the C-16-carboxylic group. Dual modified derivatives such as C-16-DMAE-amides 9 and 10 demonstrated a high antifungal activity (MICs~0.25–2 μg ml−1) comparable with AmB (1) against C. albicans, C. humicolus and A. niger. At the same time, 9 and 10 were less active than AmB (1) against F. oxysporum.
The introduction of DMAE residue into C16-moiety of 3′-N-alkyl derivative 7 did not significantly alter the activity. The double-modified derivative 12 demonstrated a slightly increased activity against F. oxysporum (MIC=1 μg ml−1) and a decreased potency against C. humicolus and A. niger (MICs=1 μg ml−1). Finally, C16-mono-modified DMAE-amide of AmB (11) showed the best activity (MICs~0.5–1 μg ml−1) close to the parent AmB (1) against all the tested bacteria.
Next, we tested whether our new compounds can be toxic for mammalian cells. Indeed, general toxicity can seriously hamper the clinical applicability of this chemical class. AmB (1) is known to be hemolytic. We therefore used AmB (1) as a reference drug in studying the ability of 3, 5, 7–12 to kill the HCT116 colon cell line or donor red blood cells. As expected, 1 was hemolytic but virtually not cytotoxic (Table 4). New derivatives were differentially active in these two test systems. Compounds 7 and 12 were more cytotoxic than 1, and 7 was even more hemolytic. In this regard, 3, 5, 8 and 9 were both nontoxic and low hemolytic; however, only 9 showed high antifungal potency (see above). Compound 11, also a promising antifungal agent, demonstrated an insignificant cytotoxicity and hemolysis (values comparable to AmB 1).
Conclusion
The chemical modification of polyene macrolide antibiotics meets serious problems due to the high MW of these compounds, poor solubility in water and organic solvents, high chemo- and photosensitivity and formation of aggregates.19 In turn, benzoxaboroles can form strong complexes with cis-diol systems, a common chemical entity found in glycopeptides, glycosides etc.20 The structure of AmB (1) also contains the cis-diol system and can form dative bonds with benzoxaboroles, thereby preventing the interaction of AmB (1) with benzoxaboroles. We succeeded in the synthesis of a series of hybrid benzoxaborole–AmB antibiotics although the yields in some cases were rather low.
In our newly synthesized series of hybrid benzoxaborole–AmB antibiotics compounds 7, 9–12 were similarly potent as the parent AmB against C. albicans, C. humicolus and A. niger. Furthermore, against F. oxysporum VKM F-140 strain, 11 and 12 showed the fungicidal activity as good as that of AmB. However, 10 and 12 were toxic for the mammalian cells, making these two derivatives disadvantageous. Thus, the introduction of the benzoxaborole residue resulted in the increase of antifungal activity of 10 and 12, however, associated with the increased cytotoxicity in comparison with AmB (1).
In 9 and 11, a potent antifungal activity was paralleled by a low toxicity for the human cells and the hemolytic activity better than (compound 9) or similar to (compound 11) AmB. The compound 9 demonstrated optimal characteristics in all the three tests, that is, antifungal activity against the three different strains, cytotoxicity and hemolytic activity. This agent was therefore prepared as a water-soluble, pharmaceutically acceptable l-Glu salt 9G potentially useful in animal models.
Experimental procedure
General
AmB (1) was purchased from Sigma-Aldrich (St Louis, MO, USA). TLC analysis was performed on the Silica gel 60 F254 plates (aluminum sheets 20 × 20 cm) Merck (Darmstadt, Germany). Column chromatography was carried out on Merck silica gel (Kieselgel G60, 0.040–0.063 mm; Darmstadt, Germany). IR spectra were recorded on a IR-Fourier spectrometer ‘Nicolet-iS10’ (detector DTGS, KBr; Thermo Scientific, Waltham, MA, USA).
NMR spectroscopy
NMR spectra were measured at 278 K in dimethyl sulfoxide-d6 (DMSO-d6) on a Bruker AVANCE 600 MHz spectrometer equipped with a triple resonance (1H, 13C and 15N) pulsed field z gradient probe (Bruker Biospin, Rheinstetten, Germany). One-dimensional NMR spectra were processed by TopSpin 2.0 and Mnova NMR (Mestrelab Research, Santiago de Compostela, Spain). 2D NMR spectra were processed by NMRPipe21 and analyzed using SPARKY.22 The 1H and 13C signal assignments were obtained from DQF-COSY and 13C-1H HSQC 2D NMR experiments. Heteronuclear experiments were measured at the natural abundance of the 13C nuclei. For the compounds 1a, 7 and 10, additionally ROESY (mixing time of 300 ms) and TOCSY (mixing time of 70 ms) 2D NMR spectra were collected.
Synthesis
C16-Amide of AmB with (S)-3-(aminomethyl)benzo[с][1,2]oxaborol-1(3Н)-ole (2)
(S)-3-(Aminomethyl)benzo[c][1,2]oxaborol-1(3Н)-ole (202 mg, 0.971 mmol) was added to a solution of AmB (1) (300 mg, 0.325 mmol) in DMSO (2 ml), pH of reaction mixture was kept at 7.5–8 by adding a solution of Et3N in DMSO. PyBOP (270 mg, 0.519 mmol) was added to the reaction mixture portionwise during 1.5 h. The reaction mixture was stirred for 4 h, and then ether was added. The mixture was stirred vigorously, then the ether layer was removed and the procedure was repeated several times until a viscous oil was formed. Acetone was added to the oil; the forming precipitate was filtered off, washed with acetone and dried in vacuum. The progress of the reactions, the components of the column and the purity of final compounds were monitored by TLC in the system CHCl3−CHCl3−MeOH−NH4OH (4:1:0.1) and HPLC analysis. The crude amide was purified by column chromatography on silica gel. The amide was dissolved in DMF (2 ml) and put on a column pre-equilibrated with CHCl3. The elution was carried out with CHCl3 (150 ml) and then with a mixture CHCl3−MeOH−NH4OH (4:1:0.1). Fractions containing the target compound were combined and evaporated to a small volume. The addition of acetone to the solution gave the precipitate that was fostered off and dried. Yield: 30 mg (10%).
3′-N-[3-(1-Hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-7-yl)propanoyl] AmB (3), 1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-6-yl)formyl AmB (4) or 1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-5-yl)formyl AmB (5)
3′-N-acyl derivatives of AmB (5–7) were prepared by the treatment of the unprotected AmB (1) with N-hydroxysuccinimide ester (-OSu) of the 3-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-7-yl)propionic acid, 1-hydroxy-1,3- dihydrobenzo[c][1,2]oxaborol-6-yl)carboxylic acid or 1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-5-yl)carboxylic acid which were obtained by the standard method using DCC. To a stirred solution of AmB (1) (150 mg, 0.162 mmol) in DMSO (100 mg ml−1) were added the solution of N-hydroxysuccinimide ester of the corresponding acid (5 eq) in the mixture of DMF-CH2Cl2 (2:1) (150 mg ml−1) and Et3N to pH 8. The reaction mixture was stirred for 12 h at room temperature, and then fivefold volume of Et2O was added. The resulting mixture was shaken intensively to partially extract DMSO and DMF, and the layer of Et2O was separated. This procedure was repeated twice. To the obtained dark yellow oil, n-BuOH (20 ml) saturated with H2O was added and the resultant solution was washed with H2O (10 ml). This procedure was repeated twice. The organic layer was evaporated in vacuum with 0.6 eq of H2O to the volume~1 ml n-BuOH solution. The organic layer was poured into a stirring mixture of Et2O to precipitate the crude reaction product. The precipitate was filtered, washed with Et2O and dried in vacuum to give a yellow powder. Crude products (5–7) were purified by the column chromatography method on silica gel in CHCl3−MeOH−H2O−HCOOH (5:1:0.01:0.01, v/v) system. The yields of the pure N-acyl-derivatives (3–5) were: 40 mg (22%) for 3, 36 mg (20.5%) for 4 and 32 mg (18%) for 5.
3′-N-(1-Hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-6-yl)sulfonyl AmB (6)
The solution of 1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-6-yl)sulfonyl chloride (35 mg, 0.16 mmol), AmB(1) (138 mg, 0.15 mmol) and Et3N (75 μl, 0.50 mmol) in the mixture of DMF: Py (1.4 ml, v/v, 1:1) was stirred at room temperature for 16 h. Then, 1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-6-yl)sulfonyl chloride (32 mg, 0.15 mmol) and Et3N (30 μl, 0.20 mmol) (pH=8) were added to the reaction mixture. The reaction mixture was stirred at room temperature for 16 h, and then fivefold volume of Et2O was added. The target compound was separated from the reaction mixture and purified by the procedure described for compounds 3–5. Yield: 27 mg (16%).
3′-N-(1-Hydroxy-1,3-dihydrobenzo[c][1,2]oxaborole-6-methyl)-AmB (7) and 3′-N,N-di(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborole-6-methyl)-AmB (8)
1-Hydroxy-1,3-dihydrobenzo[c][1,2]oxaborole-6-carbaldehyde (110 mg, 0.66 mmol) was added portionwise during 1 h to the stirred suspension of AmB (1) (200 mg, 0.22 mmol) in dry DMF (8 ml), then NaBH3CN (41 mg, 0.65 mmol) was added in three portions during 30 min. The reaction mixture was stirred at room temperature for 16 h. A mixture of acetone and ethyl ether (1:1, 30 ml) was added to the reaction mixture. The resulting precipitate was filtered off and dried. The crude precipitate (313 mg) was purified by the column chromatography method on silica gel in CHCl3−MeOH−H2O−HCOOH, 5:1:0.01:001 system. The fractions containing the target compounds 7 or 8 were combined and concentrated in vacuum. The residue was dissolved in a minimum volume of MeOH and then precipitated with the addition of ethyl ether (30 ml) to give a yellow solid which was filtered, washed with ether and dried in vacuum. Yields: 32 mg of 7 (14%) and 40 mg of 8 (15%).
C16-DMAE-amide of 3′-N-[3-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-7-yl)propanoyl] AmB (9)
Amidation of N-[3-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-7-yl)propanoyl]-AmB (3) was carried out by the method described above for the compound 2 starting from compound 3 (60 mg, 0.06 mmol) and hydrochloride of N1,N1-dimethylethane-1,2-diamine (40 mg, 0.32 mmol). The purification of crude 9 (~70 mg) was carried out by the column chromatography method on silica gel in CHCl3−MeOH− H2O−CH3COOH, (6:1:0.01:0.02) system. Yield: 35 mg (50%).
l-Glutamate salt of C16-DMAE-amide of 9 (9G)
The amide 9 was converted into DMAE salt 9G3 starting from equivalent amounts of 9 and l-Glu in the mixture of DMSO-H2O. The resulting yellow powder 9G was characterized by TLC, HPLC and NMR spectra. HPLC (Table 1) showed the presence of 9 with Rt =15.89. TLC (system BzlOH−H2O−AcOH (5:1:1, v/v) in comparison with the control samples of 9 and Glu showed the presence of both components of the salt with Rf=0.3 for 9 (yellow and UV-visible spot) and Rf=0.19 for l-Glu (ninhydrin positive spot). Also, NMR spectra of the salt 9G showed the signals of l-Glu additionally to the signals of 9 in equivalent amounts (S7).
C16-DMAE-amide of 3′-N-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-6-yl)sulfonyl-AmB (10)
Amidation of N-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-6-yl)sulfonyl-AmB (10) was carried out by the method described above for 2 starting from 6 (45 mg, 0.03 mmol) and hydrochloride of N1,N1-dimethylethane-1,2-diamine (25 mg, 0.20 mmol). The purification of crude 10 (~60 mg) was carried out by column chromatography on silica gel in CHCl3−MeOH−H2O−CH3COOH, (6:1:0.01:0.02) system. Yield: 35 mg (50%).
C16-DMAE-amide of AmB (11)
N-(2-Dimethylaminoethyl)amide of AmB (11) was obtained as described for 2 starting from AmB (1) and N1,N1-dimethylethane-1,2-diamine. The crude compound was purified by column chromatography on silica gel in system CHCl3–MeOH–H2O–NH4OH (65:30:5:1).Yield: 60%.
C16-DMAE-amide of 3′-N-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborole-6-methyl)-AmB (12)
3′-N-(2-Dimethylaminoethyl)amide of N-(1-hydroxy-1,3-dihydrobenzo [c][1,2] oxaborole-6-methyl)-AmB (12) was prepared as described for 10 starting from N-(2-dimethylaminoethyl)amide of AmB (11) and 1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborole-6-carbaldehyde. The crude compound was purified by column chromatography on silica gel in system CHCl3–MeOH–H2O–HCOOH (65:30:4:1). Yield: 25%.
Biology. Determination of in vitro antifungal, cytotoxicity and hemolytic activities
Antifungal activity
Antifungal activity (MIC values) was determined using the broth twofold microdilution method according to CLSI/NCCLS documents M27-A, M27-S3, M38-A and M38-A2,14, 15, 16, 17 (see also our previous articles).4, 18
Yeast strains C. albicans ATCC 14053, C. humicolus ATCC 9949 and the fungal strain A. niger ATCC 16404 were obtained from American Type Culture Collection Manassas, VA, USA (ATCC). F. oxysporum VKM F-140 was from Russian Collection of Microorganisms (G. K. Skryabin Institute of Biochemistry and Physiology of Microorganisms, Russian Academy of Sciences, Pushchino, Russia). RPMI 1640 medium with l-glutamine and phenol red, without sodium bicarbonate, supplemented with 0.2% glucose (ICN Biomedicals, Irvine, CA, USA) buffered with 0.165 m morpholinepropanesulfonic acid (MOPS, ACROS Organics, Geel, Belgium), pH 7.0, was used in the study. New compounds were dissolved in dimethyl sulfoxide (DMSO) at 1600 μg ml−1. Serial dilutions (down to 3.13 μg ml−1) were prepared from stock solutions in the same solvent, then diluted 50-fold in the test medium and then twice when inoculated with the microbial suspension before incubation. The final solvent concentration was 1%. MICs were measured after cultivation at 35 °C for 24 h for yeast and 48 h for fungi as the lowest concentrations of agents that prevent any visible growth.
Cell culture and cytotoxicity assays
The human colon carcinoma HCT116 cell line (ATCC) was propagated in Dulbecco modified Eagle’s medium supplemented with 5% fetal calf serum, 2 mm l-glutamine, 100 units ml−1 penicillin and 100 μg ml−1 streptomycin at 37 °C, 5% CO2 in a humidified atmosphere. The cells in logarithmic phase of growth were used in the experiments. AmB and its new derivatives were dissolved in DMSO as 10 mm stock solutions followed by serial dilutions in water immediately before the experiments. The cytotoxicity was determined in a formazan conversion assay (MTT-test). Briefly, the cells (5 × 103 in 190 μl of culture medium) were plated into a 96-well plate (Becton Dickinson, Franklin Lakes, NJ, USA) and treated with 0.1% DMSO (vehicle control) or with 10 μm of compounds 6, 8, 12–17 (0.10–50 μm; each concentration in duplicate) for 72 h. After completion of drug exposure, 50 μg of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide were added into each well for an additional 2 h. Formazan was dissolved in DMSO, and the absorbance at 540 nm was measured. The cell viability at a given drug concentration (% MTT conversion) was calculated as the percentage of absorbance in wells with drug-treated cells to absorbance in wells with DMSO-treated cells (100%).
Hemolytic activity
Erythrocytes were isolated from the peripheral blood of healthy donors after incubation at 4 °С for 2–3 h. The red blood cell suspension (100 μl) was pelleted, washed twice with saline, then the pellet was resuspended in 500 μl saline or distilled water (positive control of hemolysis). The compounds 1, 3, 5, 7–12 were added (final concentration 10 μm). The samples were incubated for 1 h at 37 °С and centrifuged at 2000 r.p.m. for 1.5 min. The OD of supernatants was measured on a Multiscan spectrophotometer FC (Thermo Scientific) at a wavelength of 540 nm. The percent hemolysis was calculated relative to positive control taken as 100%.

Synthesis of C16-benzoxaborole amide of AmB (2). AmB, amphotericin B.

Synthesis of 3′-N-substituted benzoxaborole derivatives of AmB (3–8). AmB, amphotericin B.

Synthesis of C16-DMAE-amides of 3′-N-substituted benzoxaborole derivatives of AmB (9, 10). AmB, amphotericin B; DMAE-amide, 2-(N,N-dimethylamino)ethylamide.

Synthesis of C16-DMAE-amide- (11) and C16-DMAE-amide of 3′-N-alkyl benzoxaborole derivative of AmB (12). AmB, amphotericin B; DMAE-amide, 2-(N,N-dimethylamino)ethylamide.
References
Ceregeghetti, D. M. & Carreira, E. M. Amphotericin B: 50 years of chemistry and biochemistry. Synthesis 6, 914–942 (2006).
Solovieva, S. E., Olsufyeva, E. N. & Preobrazhenskaya, M. N. Chemical modification of antifungal polyene macrolide antibiotics. Russ. Chem. Rev. 80, 103–126 (2011).
Preobrazhenskaya, M. N. et al. Synthesis and study of the antifungal activity of new mono- and di-substituted derivatives of a genetically engineered polyene antibiotic 28, 29-didehydro nystatin A1 (S44HP). Novel water-soluble derivative active in the model of the candidosis sepsis in leucopenic mice. J. Antibiot. (Tokyo) 63, 55–64 (2010).
Tevyashova, A. N. et al. Structure-antifungal activity relationships of polyene antibiotics of the Amphotericin B group. Antimicrob. Agents Chemother. 8, 3815–3822 (2013).
Adamczyk-Woźniak, A., Borys, K. M. & Sporzyński, A. Recent developments in the chemistry and biological applications of benzoxaboroles. Chem. Rev. 115, 5224–5247 (2015).
Dembitsky, V. M., Quntar, A. A. & Srebnik, M. Natural and synthetic small boron-containing molecules as potential inhibitors of bacterial and fungal quorum sensing. Chem. Rev. 111, 209–237 (2011).
Baker, S. J. et al. Discovery of a new boron containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1- benzoxaborole (AN2690), for the potential treatment of Onychomycosis. J. Med. Chem. 49, 4447–4450 (2006).
Baker, S. J., Tomsho, J. W. & Benkovic, S. J. Boron-containing inhibitors of synthetases. Chem. Soc. Rev. 40, 4279–4285 (2011).
Rock, F. L. et al. An antifungal agent inhibits an aminoacyl-Trna synthetase by trapping tRNA in the editing site. Science 316, 1759–1761 (2007).
Printsevskaya, S. S. et al. Synthesis and study of antibacterial activities of antibacterial glycopeptide antibiotics, conjugated with benzoxaboroles. Future Med Chem 5, 641–652 (2013).
Qiao, Z. et al. Chalcone–benzoxaborole hybrid molecules as potent antitrypanosomal agents. J. Med. Chem. 55, 3553–3557 (2012).
Brown, J. M. & Sidebottom, P. J. The proton magnetic resonance spectrum of amphotericin B. Tetrahedron 37, 1421–1428 (1981).
Volpon, L. & Lancelin, J.-M. Solution NMR structure of five representative glycosylated poly macrolide antibiotics with a sterol-dependent antifungal activity. Eur. J. Biochem. 269, 4533–4541 (2002).
National Committee for Clinical Laboratory Standards Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: Approved Standard M27-A, National Committee for Clinical Laboratory Standards, Wayne, PA, USA, (1997).
Clinical and Laboratory Standards Institute Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: In Third International Supplement CLSI M27-S3, Clinical and Laboratory Standards Institute, Wayne, PA, USA, (2013).
National Committee for Clinical Laboratory Standards Reference Method for Broth Dilution Antifungal Susceptibility Testing of Conidium-Forming Filamentous Fungi: Approved Standard M38-A, NCCLS, Wayne, PA, USA, (2002).
Clinical and Laboratory Standards Institute Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi: In Approved Standard, 2nd edn CLSI M38-A2 (Clinical and Laboratory Standards Institute, Wayne, PA, USA, 2008).
Preobrazhenskaya, M. N. et al. Chemical modification and biological evaluation of new semisynthetic derivatives of 28,29-didehydronystatin A1 (S44HP), a genetically engineered antifungal polyene macrolide antibiotic. J. Med. Chem. 52, 189–196 (2009).
Nicolaou, K. C. et al. Chemistry of amphotericin B. Degradation studies and preparation of amphoteronolide B. J. Am. Chem. Soc. 10, 4660–4672 (1988).
Mahalingam, A., Geononnoty, A., Balzarini, J. & Kiser, P. F. Activity and safety of synthetic lectins based on benzoboroxole-functionalized polymers for inhibition of HIV entry. Mol. Pharm. 8, 24–2475 (2011).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Goddard, T. D. & Kneller, D. G. Sparky–NMR Assignment and Integration Software (2008). http://www.cgl.ucsf.edu/home/sparky. Accessed on 06 March 2016.
Acknowledgements
We are grateful to Natalia M Malyutina (Gause Institute of New Antibiotics, Moscow) for HPLC analyses. This study was supported by the Russian Foundation for Basic Research grants 13-03-00643-a and 16-34-60110. NMR studies were partly supported by the Russian Science Foundation grant 14-14-00598.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Tevyashova, A., Korolev, A., Trenin, A. et al. New conjugates of polyene macrolide amphotericin B with benzoxaboroles: synthesis and properties. J Antibiot 69, 549–560 (2016). https://doi.org/10.1038/ja.2016.34
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2016.34
This article is cited by
-
Experimental Evaluation of Chronic Toxicity of Amphamide – a New Semisynthetic Derivative of Amphotericin B
Pharmaceutical Chemistry Journal (2020)
-
Development of a Parenteral Medicinal Formulation of the New Antifungal Semisynthetic Polyene Antibiotic Amphamide
Pharmaceutical Chemistry Journal (2020)
-
Oxaborine Derivatives: Synthesis and Therapeutic Potential
Chemistry of Heterocyclic Compounds (2020)
-
Preclinical Pharmacokinetics Study of Amphamide: a New Semisynthetic Antifungal Antibiotic
Pharmaceutical Chemistry Journal (2020)
-
Synthesis and evaluation of biological activity of benzoxaborole derivatives of azithromycin
The Journal of Antibiotics (2019)
