
Abstract
Complex polyketides comprise a large number of natural products that have broad application in medicine and agriculture. They are produced in bacteria and fungi from large enzyme complexes named type I modular polyketide synthases (PKSs) that are composed of multifunctional polypeptides containing discrete enzymatic domains organized into modules. The modular nature of PKSs has enabled a multitude of efforts to engineer the PKS genes to produce novel polyketides of predicted structure. We have repurposed PKSs to produce a number of short-chain mono- and di-carboxylic acids and ketones that could have applications as fuels or industrial chemicals.
Similar content being viewed by others
Introduction
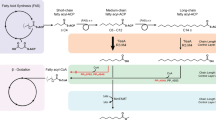
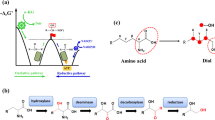
Complex polyketides represent a family of natural products that possess a wide variety of pharmacological or biological activities. Numerous polyketides and their semisynthetic derivatives have been approved for clinical use in humans or animals, including the antibiotics erythromycin, tylosin and tiacumicin B, the antifungal agents amphotericin B and nystatin, the immunosuppressants rapamycin and FK506, the antiparasitic agent avermectin, the coccidiostats monensin and salinomycin and the insecticide family of spinosyns, to name a few, with annual sales exceeding $20B USD.1 Many others have antitumor activity and several are currently in clinical trials. As can be seen in Figure 1, these compounds share very little structural similarity. What unites them is their common mechanism of biosynthesis. These compounds are produced by a class of enzymes called type I modular polyketide synthases (PKSs) that condense and reduce a small series of acyl-CoA precursors (for example, propionyl-CoA, malonyl-CoA, methylmalonyl-CoA) to build the polyketide backbone in a step-wise manner that resembles fatty acid biosynthesis. PKSs are composed of modules, each of which is responsible for the addition of 2 carbon (C) atoms to the growing acyl chain. Each module is composed of a series of enzymatic or structural domains that select the extender CoA unit, condense it to the growing chain and reduce the resultant β-carbonyl formed to the extent determined by the functional reducing domains present within the module. As shown in Figure 2, which displays schematic representations for the biosynthesis of two complex polyketides,2, 3 the complete structure of a complex polyketide is programmed by its cognate PKS, by the number and composition of modules.

Structures of complex polyketides. Atoms shown in black are programmed by polyketide synthases. Lighter colored atoms are programmed by enzymes involved in post-polyketide tailoring reactions (see Figure 2).

Biosynthesis of erythromycin (a) and tiacumicin (b). Schemes for the step-wise biosynthesis of the polyketide backbones of 6-deoxyerythronolide B (6-dEB) and tiacumicinone showing the structure of the intermediate tethered to the acyl carrier protein (ACP) after all reactions by the modular domains are complete. In each module the domains and the chemical reactions they determine are as follows: acyltransferase (AT) domains specify the CoA substrate employed by binding it and transferring it the ACP (not shown) via a thioester linkage; ketosynthase (KS) domains carry out decarboxylative condensation employing the acyl chain that has been transferred to it from the upstream ACP and the substrate on the ACP domain, generating a 3-keto acyl ACP intermediate (not shown); ketoreductase (KR) domains stereospecifically reduce the 3-keto group and they also determine the stereochemistry of the 2-alkyl (methyl, ethyl, etc.) group, if present; dehydratase (DH) domains remove the 2,3-H+ and OH− leaving a 2,3-double bond; enoylreductase (ER) domains reduce the double bond leaving a 3-methylene; and thioesterase (TE) domains release the completed acyl chain from the polyketide synthase (PKS) by producing a macolactone, in the two cases shown here, or via hydrolysis as shown in Figure 3 to produce a free carboxylic acid. 6-dEB and tiacumicinone are converted to erythromycin and tiacumicin B, respectively, through the action of various specific hydroxylases, glycosylases, O-methyltransferases, acyltransferases, etc. (not shown).
Since the concept of modular polyketide biosynthesis was initially report by Katz and colleagues2 more than 25 years ago, numerous attempts have been made to ‘reprogram’ PKSs to produce altered polyketide backbones with improved or novel pharmaceutical activities.4 Although many examples of novel compounds from genetically engineered PKSs have been reported, most efforts have either failed outright or have resulted in reduced titers of product generated as to make the testing of the novel compounds impractical. It is clear that not enough is currently known about the structural requirements of domains and modules for optimum biochemical activity, including acyl chain passage from one module to the next to enable effective PKS reprogramming.
Our work, located at the Joint BioEnergy Research Institute, a US Department of Energy-funded facility, centers on repurposing type I modular PKSs to produce a variety of short-chain compounds that can be useful as biofuels or industrial chemicals. Our standardized approach is to first design and construct the target PKS gene(s) to generate the desired compound, produce the soluble PKS protein(s) in Escherichia coli and use the purified material to demonstrate production of detectable levels of the desired compound in vitro. These enzyme systems would also be employed to determine the kinetics, the relative specificities of the required substrates, etc. (Yuzawa et al.5 reviewed in the paper dedicated to Professor David Cane). The next steps would be to demonstrate production of the targeted compound at commercially feasible titers. Our efforts in this area over the past 5 years are described in this review.
Opportunities
PKS engineering to produce fuels
Transportation fuels, in general, are highly reduced hydrocarbons of varying chain length that are burned aerobically in transportation vehicle engines to power movement, with the resultant production of one molecule of CO2 for every atom of carbon utilized. The current design of engines requires the production of three types of fuels, all generated from the refining of crude petroleum: gasoline (petrol) for cars, aviation fuel for planes and diesel for a variety of heavy and light vehicles. Gasoline is a mixture of molecules composed of small hydrocarbons (5–8 C atoms) containing mainly straight- and branched-chain alkanes and cycloalkanes, and a small amount of aromatic compounds. Short-chain alcohols, ethanol and isobutanol, can also be added to gasoline mixtures. An important component of gasoline is isooctane, 2,2,4-trimethylpentane, that is assigned an octane rating of 100, and against which all fuels are rated. Octane rating is based on the ability to prevent engine knock (ignition of a separate pocket of an air–fuel mixture that does not provide energy to push the piston) in a spark-ignition engine. The high octane rating of isooctane is because of the density of the molecule, attributed to the significant degree of branching of the carbon backbone. Jet fuel is mainly composed of kerosene (alkyl-aromatics, 12–15 C atoms), but also contains heptane and isooctane. Petroleum-based diesel is composed of saturated straight-, branched- and cyclic-alkanes (75%), and aromatics (25%: napthalenes and alkyl-benzenes), with an average size ranging from 12 to 15 C atoms.
Although PKS modules can be refactored to generate compounds of virtually any length, with the required carbon length and extents of both branching and saturation for any of the three fuels, there have been great advances in employing the enzyme machinery of isoprenoid and fatty acid biosynthesis for the bio-based production of both jet fuels and diesel, respectively.6 We have focused, therefore, on employing PKSs to make bio-based gasoline replacements. It should be noted that there are no published examples of PKSs producing nonoxygenated molecules. On the other hand, generation of a bio-based acid or alcohol from an engineered PKS that, when chemically reduced (via hydrogenation), results in the molecule identical in structure to a petroleum-based compound may be a viable route to a biofuel. Our goal, therefore, was to design PKS systems to produce the compounds 2,4,4-trimethyl-3-hydroxypentanoic acid or 2,2,4-trimethyl-3-hydroxypentanoic acid that, when chemically reduced, would be converted to isooctane.
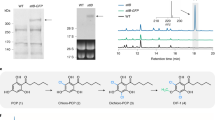
2,4,4-Trimethyl-3-hydroxypentanoic acid can be generated from a refactored type I modular PKS that programs a single condensation reaction between the starter pivaloyl-CoA and the extender methylmalonyl-CoA followed by reduction of the newly generated β-keto group and hydrolysis of the final polyketide intermediate attached to the enzyme. Our initial efforts to produce 3-hydroxy acids began with repurposing the avermectin PKS that produces the 16-membered macrocyclic lactone, 6,8a-seco-6,8a-deoxy-5-oxoavermectin aglycon.7 The loading didomain of the avermectin PKS has been shown to use a huge variety of starter acyl-CoA substrates to initiate the biosynthesis by AVES1,8 and it was hoped that it would also use pivaloyl-CoA as a starter. AVES1 is composed of the loading acyl transferase–acyl carrier protein (AT-ACP) didomain, module 1 (KS-AT-KR-ACP) and module 2 (KS-AT-DH-KR-ACP). To produce the 3-hydroxy acid, we inserted the erythromycin PKS thioesterase (TE) domain downstream of module 1 and introduced a stop codon to terminate translation. Unfortunately, the resulting hybrid PKS (AT-ACP-KS-AT-KR-ACP-TE) was not solubly expressed in E. coli. Hence, we turned to the lipomycin PKS, whose loading acyltransferase (AT) domain shares 50% amino acid similarity with the avermectin loading AT.9 Unlike the avermectin PKS, the first protein of the lipomycin PKS (LipPks1) contains only the loading didomain and module 1, and linkage of LipPks1 to the erythromycin PKS TE domain gave rise to the soluble and enzymatically active hybrid PKS LipPks1+TE (AT-ACP-KS-AT-KR-ACP-TE) in E. coli (Figure 3). Unfortunately, although we could detect a small amount of product with mass corresponding to 2,4,4-trimethyl-3-hydroxypentanoic acid by LC-MS when pivaloyl-CoA was used as substrate, extensive biochemical analysis showed that the kcat/KM value with this starter was less than 1/50 of the kcat/KM values determined when the natural starters, isobutyryl-CoA (α-lipomycin) or 2-methylbutyryl-CoA (21-methyl-α-lipomycin),10 were employed. The products, 2,4-dimethyl-3-hydroxypentanoic acid and 2,4-dimethyl-3-hydroxyhexanoic acid, can be converted to 2,4-dimethylpentane and 2,4-dimethylhexane using chemo-, bio- and integrated catalytic approaches11 that could ultimately be identified as bio-based substitutes for gasoline.

Repurposing α-lipomycin biosynthetic pathway to produce isooctane precursors. Thioesterase (TE) from the erythromycin polyketide synthase (PKS; black box) was attached to the C-terminus of LipPks1 to release the product (top). The resulting engineered PKS (LipPks1+TE) produces 2,4,4-trimethyl-3-hydroxypentanoic acid from pivaloyl-CoA and methylmalonyl-CoA in the presence of NADPH. cMT (black box)-inserted LipPks1+TE may be capable of producing 2,4,4-trimethyl-3-hydroxypentanoic acid (bottom). Both acid products can be chemically converted into isooctane. cMT, C-methyltransferase; all other abbreviations as in Figure 2.
The PKS design to produce 2,2,4-trimethyl-3-hydroxypentanoic acid is also shown in Figure 3. The proposed hybrid PKS would consist of a hybrid PKS with the following domains: AT-ACP-KS-AT-cMT-KR-ACP-TE, where cMT is a C-methyltransferase. The biosynthesis would employ isobutyryl-CoA as the starter. The lipomycin loading AT-ACP didomain would fill this role. The second AT domain would be matched with the cMT domain so that if the AT specifies malonyl-CoA, the cMT employed would be capable of carrying out dimethylation. Alternatively, the AT could specify methylmalonyl-CoA and the cMT domain would only need to catalyze a single methylation. The only example of a bacterial PKS module composed of KS-AT-cMT-KR-ACP domains that could generate a gem-dimethyl-containing product is found in the yersiniabactin PKS (designated HMWP1) from the Gram-negative bacterium Yersinia pestis, where the AT domain specifies malonyl-CoA as a substrate and the cMT domain catalyzes dimethylation.12 However, attempts to link the lipomycin loading didomain to HMWP1 to produce soluble active protein were not successful. An alternative approach is to introduce a cMT domain into, for example, LipPks1+TE. However, to date, no report of successful construction of a module containing a heterologous cMT domain has been reported.
PKS engineering to produce industrial chemicals
Industrial chemicals comprise hundreds of compounds that have commercial applications in virtually every aspect of modern life. They are divided into two broad categories: commodity chemicals that have low unit value but are produced and used in very high quantities (for example, ca. 2 billion tons of adipic acid are produced annually) and specialty chemicals that have moderate to high market value but are produced at modest quantities. Examples of commodity chemicals include monomers (for example, adipic acid) used to make polymers (for example, nylon, polypropylene, polyvinyl chloride, plastics) or synthetic rubber, and a variety of chemicals used as surfactants, adhesives, expanders, paint additives, etc. Specialty chemicals include those used as pesticides, fragrances, flavors, food additives and disinfectants, or chemicals used for textiles, specialty coatings, printing or paper production, to name a few. In 2015, the specialty chemical market in North America exceeded $153B USD (http://www.statista.com/statistics/548491/market-value-of-the-specialty-chemical-industry-in-north-america/). A large fraction of industrial chemicals come from petroleum, the remainder come from natural gas, plants or inorganic sources. Industrial chemicals as a group are produced from not more than 15% of the total amount of petroleum refined, and thus the motivation to replace petroleum as the source of these compounds is not driven by the limitation of supply but rather by the volatility of the price of oil, as well as by environmental, health or safety concerns that apply to the current methods used for their production.
In contrast to bio-based production of fuels, which offers a large opportunity for novel compounds to be generated, all published current efforts at producing bio-based chemicals are directed toward ‘drop-in’ replacements of existing products. Our efforts in this area are focused on designing PKS systems to produce either drop-in replacements or novel chemicals with new or improved properties.
3-Hydroxy acids
In contrast to the proposed role of serving as bio-based intermediates in the chemical production of isooctane or other gasoline components, as described above, 3-hydroxy acids are currently employed as monomers for the production of biodegradable polyesters. Examples include lactic acid for the chemical production of polylactic acid, and 3-hydroxybutyrate, 3-hydroxyvalerate and 3-hydroxyhexanoate for the biological production of polyhydroxybutyrate, polyhydroxyvalerate and polyhydroxyhexanoate, respectively. Application of these polymers include food wraps, plastic trays, drink bottles, heat seal resins, heart valves and fibers in clothing, wipes and blankets. Many of these polymers are readily biodegraded, making them ideal replacements for many petroleum-based polymers that cannot be biodegraded.
Engineered PKSs may provide further diversity in the production of monomers that have not been reachable using traditional synthetic chemistry. As described above in the description of biofuels, a key finding in our in vitro work was the broad substrate specificity of the loading AT of the lipomycin PKS.13 The loading AT accepted all of starter acyl-CoAs tested except for acetyl-CoA (propionyl-CoA, n-butyryl-CoA, isobutyryl-CoA, 2-methylbutyryl-CoA, isovaleryl-CoA, and, to a much lesser degree, pivaloyl-CoA) to produce C6-C8 2-methyl-3-hydroxy acids with methylmalonyl-CoA in the presence of NADPH (Figure 4a).

Repurposing antibiotic-producing type I modular polyketide synthases (PKSs) to produce industrial chemicals. (a) LipPks1+TE produces 3-hydroxy carboxylic acids from various starter CoAs and methylmalonyl-CoA in the presence of NADPH (left). AT-swapped LipPks1+TE produces 3-hydroxy carboxylic acids from various starter CoAs and malonyl-CoA in the presence of NADPH (right). (b) KR-inactivated LipPks1+TE produces ethyl ketones from various starter CoAs and methylmalonyl-CoA (left). KR-inactivated, AT-swapped LipPks1+TE produces methyl ketones from various starter CoAs and malonyl-CoA (right). (c) KR in BorA2 was replaced with DH from BorA3 (dotted box) and KR and ER from various PKS modules (boxes shown in black). TE from the erythromycin PKS (black box) was attached to the C-terminus of BorA2 to release the product. The resulting KR-swapped PKS produces adipic acid from succinyl-CoA and malonyl-CoA in the presence of NADPH. Abbreviations are shown in Figure 2.
The second AT, located in module 1 of LipPks1+TE, is specific for methylmalonyl-CoA. AT domain swapping is the most common approach to change the extender substrate employed in a module in type I modular PKSs.14 Recently, we have systematically analyzed the segments of AT domains and associated linkers in AT exchanges in vitro using two monomodular PKS systems, including LipPks1+TE as models, and have identified an AT domain boundary that can be used to replace methylmalonyl-specifying AT domains with those that specify malonyl-CoA while maintaining protein stability and module activity. We have used these modified LipPKS1+TE constructs to produce a number of 2-desmethyl-3-hydroxy acids.15 Importantly, the optimized domain boundary is highly conserved that facilitates AT domain replacements in many type I PKS modules. Recent advances in genome sequencing and the study of crotonyl-CoA carboxylase/reductase homologs identified many extender substrates and their corresponding AT domains in nature.16, 17 At least 20 acyl-CoA substrates, all malonyl-CoA analogs, have been found to be incorporated into naturally occurring polyketides, and many of the corresponding AT domains have been identified in sequenced PKSs. These acyl-CoAs include long-chain alkylmalonyl-CoAs, halogenated malonyl-CoAs and benzylmalonyl-CoA. Using the optimized AT domain boundary, we may be able to combine various starter and extender acyl-CoAs to producing a larger variety of 3-hydroxy acids.
Type I modular PKS products are typically rich in stereocenters and altering the stereochemistry is an important method to increase chemical diversity. The exchange of ketoreductase (KR) domains in model PKSs has been shown to predictably alter the stereochemistry of both the β-hydroxyl and α-methyl groups.18, 19, 20 Using LipPks1+TE as a model, we could successfully alter the 2-methyl stereochemistry from S to R that provided the first experimental evidence of stereochemical conversion of polyketide products from anti to syn in PKS engineering experiments.20 Although we could not change the β-hydroxy stereochemistry by KR swapping in our model PKS, Leadlay and co-workers19 had successfully altered the stereochemistry of a β-hydroxy group from S to R by exchanging a KR domain in a different model PKS. These observations clearly suggest that our current KR engineering knowledge is not sufficient. However, we believe that further efforts will be made and the resulting knowledge will allow us to control stereochemistries of type I modular PKS products in a highly efficient manner. This would have important consequences for the bio-based production of polyhydroxyalkanoates that currently employs the enzyme polyhydroxyalkanoate synthase to polymerize 3R-hydroxy acids.21 The variety of polymers observed is currently limited by the variety of 3R-hydroxy acids produced in vivo. Expanding the number and variety of 3R-hydroxy acids by employing refactored modular polyhydroxyalkanoates, as described here, can enable the bio-based production of novel polyhydroxyalkanoates.
Ketones
Short-chain alkyl ketones such as 2-butanone and 4-methyl-2-pentanone are industrially important solvents that are used in a variety of applications, including paints, coatings, adhesives, magnetic tapes, inks as well as for cleaning and extraction. Although some microbes (mainly Clostridium sp.) naturally produce acetone, no microbe or metabolic pathway, to our knowledge, is known to produce detectable levels of other short-chain alkyl ketones.
3-Keto acids are chemically unstable and known to decompose to ketones spontaneously under mild conditions.20, 22, 23, 24 As described above, we have produced a variety of 3-hydroxy acids by repurposing the LipPks1+TE. Using the same PKS but inactivating the KR function, we could successfully produce five ethyl ketones from their corresponding 3-keto acids using propionyl-CoA, n-butyryl-CoA, isobutyryl-CoA, 2-methylbutyryl-CoA or isovaleryl-CoA as the starter substrate.20, 23 These ethyl ketones contain 5 to 7 C atoms (Figure 4b). In order to produce other alkyl ketones, we replaced the AT domain in module 1 to change the extender substrate from methylmalonyl-CoA to malonyl-CoA. The resulting AT-swapped PKS was capable of producing five methyl ketones in the C4 to C6 range including 2-butanone and 4-methyl-2-pentanone.15 If the KS were sufficiently substrate agnostic in the chain elongation reaction, it would be possible to produce C4–C13 ketones with different branching patterns and keto group positions by condensing various lengths of starter and extender substrates. Medium-chain alkyl ketones (C11–C15) are probably neither good solvents nor good gasoline alternatives but could be relevant to the jet fuel, diesel or flavor and fragrance industries.
Diacids
Dicarboxylic acids (diacids) are used as a monomer component in the production of polyamides and polyesters. For example, Nylon 6,6 is produced by polymerization of adipic acid, with its chemically derived analog hexane-1,6-diamine. Nylon is widely used in our society as fibers (apparel, carpeting and rubber reinforcement), shapes (car parts and electrical equipment) and in films (mostly for food packaging). Although current diacids that are used in polymer materials are restricted to symmetric, linear (mostly C6, C10 and C12) or aromatic (terephthalate) diacids, engineered type I modular PKSs may be able to produce diacids of different lengths and containing various side chains.
Adipic acid can be produced by a single condensation reaction between the starter succinyl-CoA and the extender malonyl-CoA, followed by full reduction of the resulting β-keto group and hydrolysis of the final polyketide intermediate. Recently, as shown in Figure 4c, we have demonstrated that BorA1 of the borreledin PKS, which contains the loading AT-ACP didomain, could incorporate (load and transfer to the ACP domain) succinyl-CoA.25 The presumed natural substrate of the loading AT is trans-1,2-cyclopentanedicarboxylic acid (trans-1,2-CPDA) activated to its cognate CoA.26 We have also demonstrated that the downstream protein BorA2 (KS-AT-KR-ACP) could accept the succinyl starter and extend with malonyl-CoA to produce 3-hydroxyadipic, attached to the ACP in BorA2.25 This enzyme-bound product was released as a 3-hydroxyadipic acid by linking the erythromycin PKS TE domain immediately downstream of BorA2.27
As BorA2 carries only a KR domain, production of adipic acid required engineering of BorA2 to carry the full complement of reductive domains: KR, dehydratase (DH) and enoyl reductase (ER) domains. Hence, we replaced the native KR domain of BorA2 with a contiguous segment containing the KR-DH-ER tridomains from a number of PKS modules, and subsequently introduced additional replacements of the DH domain. Interestingly, we found that the DH domain from the adjacent downstream module of the borrelidin PKS (BorA3) greatly enhanced production of adipic acid in vitro.27 We speculate the presence of the terminal carboxyl on the acyl chain undergoing dehydration might negatively influence binding to DH domains that do not normally employ substrates with terminal carboxyls, whereas the native substrate of the DH domain of BorA3 carries a terminal carboxyl group. Going beyond adipic acid, further swapping of the AT domain may enable us to produce adipic acid analogs carrying side chains that have a terminal double bond, a chloro- or a keto group, each of which can be used to crosslink polymer chains, giving the polymers unique properties that are useful in industrial applications.
Challenges
PKS engineering
The prospect of using type I modular PKSs to produce bio-based fuels and industrial chemicals faces two major challenges. The first challenge is to maximize the biochemical activity of modules and domains in hybrid PKS systems. Currently, we can change a substrate introduced by a PKS module as well as the stereochemistries of polyketide intermediates produced by AT and KR domain swapping, but replacing an entire catalytic domain in a module may significantly change and/or destabilize the module structure that would compromise all the catalytic functions of the engineered PKS. Although we have successfully optimized domain boundaries for AT domain swapping, similar efforts are needed to optimize other domain swapping methods, as well as introducing new domains (for example, MT) into heterologous modules.
An important issue facing the activity of hybrid PKSs is the efficiency of transfer of the nascent acyl chain between the ACP of the upstream module to the KS domain of its noncognate partner. Many KS domains have stringent substrate specificities resulting in either the failure or the reduced efficiency of completing synthesis of the polyketide. In addition, many biochemical studies have indicated that the rate-determining step in type I modular PKSs is the polyketide chain elongation reaction, suggesting a key role for the KS domain in (extender) substrate recognition. This would become important when AT swaps are introduced into a module and the KS sees a novel substrate on its cognate ACP. Both more sophisticated KS engineering (for example, KS domain or subdomain swapping) and bioinformatics approaches to predict KS substrate specificities need to be developed to tackle the important issue of KS gatekeeping in order to efficiently generate target compounds by hybrid PKSs.
Production host
The larger challenge is whether it is possible to produce the desired compound at economically viable costs. Production costs for biofuels and commodity chemicals are determined largely by the cost of the media ingredients (for example, sugars, amino acids, etc.) and the length of time of time necessary to achieve maximum titer of product. Hence, margins are determined by titers achieved, rates of production and yields (grams of product to grams of carbon source employed). Additional costs are incurred in the recovery and purification of the product.
What is the ‘best’ host for production of type I modular PKS-based biofuels and chemicals? Streptomycetes, filamentous soil bacteria, are very well-known producers of drugs produced by type I modular PKSs. The engineered PKSs employed to produce the compounds described in this review all originate in various Streptomyces strains. Industrial Streptomyces strains (generated from many years of classical strain improvement efforts) can produce 10–100 g l−1 of polyketide drugs from naturally encoded PKSs after extended (7–10 days) fermentation in rich media (R Baltz personal communication); the basis of high titer production is only poorly understood. Several Streptomyces hosts, such as Streptomyces coelicolor, Streptomyces lividans, Streptomyces albus, Streptomyces fradiae and Streptomyces avermitilis have been used as heterologous hosts for polyketide production encoded by various type I modular PKSs, although titers achieved in unoptimized shake flask fermentations are only in the 1–50 mg l−1 range.28 Although E. coli and yeast have also been used as heterologous hosts, most probably because of the availability of well-characterized genetic tools, these hosts are likely suboptimal because they are phylogenetically distant from Streptomyces. Heterologous expression of large and GC-rich type I modular PKS genes may require machinery native to Streptomyces to stably generate the corresponding mRNAs and proteins. Streptomyces may also have specific chaperone systems to properly fold large proteins into their active forms. It is also well known that they natively produce many acyl-CoAs, although some polyketide biosynthetic clusters also carry sets of genes to make specialized acyl CoAs required by the cognate PKS. E. coli and yeast usually produce malonyl-CoA only.
Standard, high-titer fermentations of polyketide drugs in Streptomyces require very high quantities of carbohydrate (glucose, starch) and protein, most of which is not incorporated into product, as well as, in some cases, expensive elixers such as corn steep liquor and oils. Once conditions are established, companies seek only titer improvements to increase profitability. Clearly, for low-value fuels and drop-in replacement commodity chemicals, which must compete with their petroleum based-counterparts, current PKS-employed fermentation conditions are not industrially viable. Efforts to increase production rates, increase titers and yields in less complex, less expensive media must be undertaken.
In general, the high polyketide producing Streptomyces strains that exist have been engineered to produce a single, native product. Whether they can also serve as hosts for the production of other polyketides remains to be seen. Unfortunately, as they are the property of private companies and are not available to academic (or non-company) researchers, we are left with the formidable challenge of developing strains that are currently low-level producers into high-level producers of multiple products. Because of a large gap in our understanding, straightforward approaches to achieve this are not known. Recent technical advances in genome sequencing has enabled the sequencing of several hundred Streptomyces genomes, revealing the presence of an average of 25 secondary metabolite gene clusters per genome, many of which are encoded by type I modular PKSs. It is not known how many of these clusters are expressed or how many new compounds are produced. Will the biosynthesis of these native compounds compete for precursors with the targeted fuels or chemicals being produced? Is it necessary to delete these competing pathways from the host? Moreover, is the pool of precursors and supply of NADPH required for the biosynthesis of the desired compound, sufficient to support high titer production? The application of metabolomics employing MS analysis can answer many of these questions. In addition, many new gene editing tools for Streptomyces have been developed recently to enable the rapid removal or addition of genes from the chromosome. A number of actinophage based site-specific integrase systems have been developed to enable the introduction of PKS genes into various cognate attB sites in the genome, and hybrid PKS genes can be easily synthesized, assembled in yeast, transferred to E. coli and introduced into Streptomyces via conjugation. Recently, many synthetic and natural promoters were developed for use in various Streptomyces strains.29, 30, 31, 32 Luo, Zhang and coworkers33, using a fluorescence-activated cell sorting-based high-throughput screening with a fluorescent protein reporter, characterized ca. 200 native or synthetic promoters and ca. 200 ribosomal-binding sites in Streptomyces albus, and identified many promoters and ribosomal-binding sites that are much stronger than the well-characterized ermE promoter from Saccharopolyspora erythraea. RNA-Seq and MS-based targeted proteomics34 can be used to directly examine rates of expression of PKS genes and rates of PKS protein production in Streptomyces hosts. Finally, CRISPR-Cas9 systems have been developed for genome editing of Streptomyces strains.35, 36, 37 Efficient generation of gene deletions were demonstrated in S. coelicolor, S. lividans, S. albus, Streptomyces viridochromogenes and Streptomyces pristinaespiralis. This promising tool should also be tested for the efficacy of introducing single or multiple insertions in various Streptomyces hosts.
Conclusions
Polyketides and their semisynthetic analogs are exquisite drugs. The complexity of their structure allows them to fit into the proteins or protein complexes they target with very high precision and with little off-target activity. Small, redesigned segments of these compounds can be produced by refactoring the polyketide biosynthetic machinery, the polyketide synthases. These compounds lack the architectural elegance of the larger drugs but can have properties as fuels or chemicals. These compounds are not designed to interact with targets but to generate useful energy when burned, to be polymerized into larger materials or to be used as solvents for industrial purposes, none of which are unique to these or any other class of molecules. We are of the opinion, however, that in the long term, yet to be exploited value of polyketides in the chemical industry will come from precisely the structural uniqueness of the resulting compounds that will impart useful properties not currently available in chemicals obtained from petroleum or from other biological pathways. The challenge is to make these at reasonable cost.
References
Weissman, K. J. Introduction to polyketide biosynthesis. Methods Enzymol. 459, 3–16 (2009).
Donadio, S, Stave, M. J., McAlpine, J. B., Swanson, S. J. & Katz, L. Modular organization of genes required for complex polyetide biosynthesis. Science 252, 675–679 (1991).
Xiao, Y. et al. Characterization of tiacumicin B biosynthetic gene cluster affording diversified tiacumicin analogues and revealing a tailoring dihalogenase. J. Am. Chem. Soc. 133, 1092–1105 (2011).
Yuzawa, S., Kim, W., Katz, L. & Keasling, J. D. Heterologous production of polyketides by modular type I polyketide synthases in Escherichia coli. Curr. Opin. Biotechnol. 23, 727–735 (2012).
Yuzawa, S., Keasling, J. D. & Katz, L. Insights into polyketide biosynthesis gained from repurposing antibiotic-producing polyketide synthases to produce fuels and chemicals. J. Antibiot. (Tokyo) 69, 494–499 (2016).
Beller, H. R., Lee, T. S. & Katz, L. Natural products as biofuels and bio-based chemicals: fatty acids and isoprenoids. Nat. Prod. Rep. 32, 1508–1526 (2015).
Ikeda, H., Nonomiya, T., Usami, M., Ohta, T. & Omura, S. Organization of the biosynthetic gene cluster for the polyketide anthelmintic macrolide avermectin in Streptomyces avermitilis. Proc. Natl Acad. Sci. USA 96, 9509–9514 (1999).
Moore, B. S. & Hertweck, C. Biosynthesis and attachment of novel bacterial polyketide synthase starter units. Nat. Prod. Rep. 19, 70–99 (2002).
Bihlmaier, C. et al. Biosynthetic gene cluster for the polyenoyltetramic acid alpha-lipomycin. Antimicrob. Agents Chemother. 50, 2113–2121 (2006).
Yuzawa, S., Eng, C. H., Katz, L. & Keasling, J. D. Enzyme analysis of the polyketide synthase leads to the discovery of a novel analog of the antibiotic alpha-lipomycin. J. Antibiot. (Tokyo) 67, 199–201 (2014).
Deneyer, A. et al. Alkane production from biomass: chemo-, bio- and integrated catalytic approaches. Curr. Opin. Chem. Biol. 29, 40–48 (2015).
Poust, S. et al. Divergent mechanistic routes for the formation of gem-dimethyl groups in the biosynthesis of complex polyketides. Angew. Chem. Int. Ed. Engl. 54, 2370–2373 (2015).
Yuzawa, S., Eng, C. H., Katz, L. & Keasling, J. D. Broad substrate specificity of the loading didomain of the lipomycin polyketide synthase. Biochemistry 52, 3791–3793 (2013).
Dunn, B. J. & Khosla, C. Engineering the acyltransferase substrate specificity of assembly line polyketide synthases. J. R. Soc. Interface 10, 20130297 (2013).
Yuzawa, S. et al. Comprehensive in vitro analysis of acyltransferase domain exchanges in modular polyketide synthases and its application for short-chain ketone production. ACS Synth. Biol. (e-pub ahead of print 6 September 2016; doi:10.1021/acssynbio.6b00176).
Chang, C. et al. Uncovering the formation and selection of benzylmalonyl-CoA from the biosynthesis of splenocin and enterocin reveals a versatile way to introduce amino acids into polyketide carbon scaffolds. J. Am. Chem. Soc. 137, 4183–4190 (2015).
Wilson, M. C. & Moore, B. S. Beyond ethylmalonyl-CoA: the functional role of crotonyl-CoA carboxylase/reductase homologs in expanding polyketide diversity. Nat. Prod. Rep. 29, 72–86 (2012).
Annaval, T., Paris, C., Leadlay, P. F., Jacob, C. & Weissman, K. J. Evaluating ketoreductase exchanges as a means of rationally altering polyketide stereochemistry. Chembiochem. 16, 1357–1364 (2015).
Kellenberger, L. et al. A polylinker approach to reductive loop swaps in modular polyketide synthases. Chembiochem. 9, 2740–2749 (2008).
Eng, C. H. et al. Alteration of polyketide stereochemistry from anti to syn by a ketoreductase domain exchange in a type i modular polyketide synthase subunit. Biochemistry 55, 1677–1680 (2016).
Stubbe, J. et al. Nontemplate-dependent polymerization processes: polyhydroxyalkanoate synthases as a paradigm. Annu. Rev. Biochem. 74, 433–480 (2005).
Goh, E. B., Baidoo, E. E., Keasling, J. D. & Beller, H. R. Engineering of bacterial methyl ketone synthesis for biofuels. Appl. Environ. Microbiol. 78, 70–80 (2012).
Yuzawa, S., Katz, L. & Keasling, J. D. Producing 3-hydroxycarboxylic acid and ketone using polyketide synthase. US Patent Application 20150307855 (2015).
Goh, E. B. et al. Substantial improvements in methyl ketone production in E. coli and insights on the pathway from in vitro studies. Metab. Eng. 26, 67–76 (2014).
Hagen, A. et al. In vitro analysis of carboxyacyl substrate tolerance in the loading and first extension modules of borrelidin polyketide synthase. Biochemistry 53, 5975–5977 (2014).
Olano, C. et al. Biosynthesis of the angiogenesis inhibitor borrelidin by Streptomyces parvulus Tu4055: cluster analysis and assignment of functions. Chem. Biol. 11, 87–97 (2004).
Hagen, A. et al. Engineering a polyketide synthase for in vitro production of adipic acid. ACS Synth. Biol. 5, 21–27 (2016).
Baltz, R. H. Streptomyces and Saccharopolyspora hosts for heterologous expression of secondary metabolite gene clusters. J. Ind. Microbiol. Biotechnol. 37, 759–772 (2010).
Bai, C. et al. Exploiting a precise design of universal synthetic modular regulatory elements to unlock the microbial natural products in Streptomyces. Proc. Natl Acad. Sci. USA 112, 12181–12186 (2015).
Wang, W. et al. Development of a synthetic oxytetracycline-inducible expression system for streptomycetes using de novo characterized genetic parts. ACS Synth. Biol. 5, 765–773 (2016).
Seghezzi, N., Amar, P., Koebmann, B., Jensen, P. R. & Virolle, M. J. The construction of a library of synthetic promoters revealed some specific features of strong Streptomyces promoters. Appl. Microbiol. Biotechnol. 90, 615–623 (2011).
Siegl, T., Tokovenko, B., Myronovskyi, M. & Luzhetskyy, A. Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab. Eng. 19, 98–106 (2013).
Luo, Y., Zhang, Y., Barton, K. W. & Zhao, H. Systematic identifaction of a panel of strong constitutive promoters from Streptomyces albus. ACS Synth Biol 4, 1001–1010 (2015).
Batth, T. S., Keasling, J. D. & Petzold, C. J. Targeted proteomics for metabolic pathway optimization. Methods Mol. Biol. 944, 237–249 (2012).
Cobb, R. E., Wang, Y. & Zhao, H. High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth. Biol. 4, 723–728 (2015).
Tong, Y., Charusanti, P., Zhang, L., Weber, T. & Lee, S. Y. CRISPR-Cas9 based engineering of actinomycetal genomes. ACS Synth. Biol. 4, 1020–1029 (2015).
Huang, H., Zheng, G., Jiang, W., Hu, H. & Lu, Y. One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta Biochim. Biophys. Sin. (Shanghai) 47, 231–243 (2015).
Acknowledgements
This work was supported by the Joint BioEnergy Institute that is funded by the Office of Science, Office of Biological and Environmental Research of the US Department of Energy (Contract No. DE-AC02-05CH11231), by the Department of Energy, ARPA-E Electrofuels Program (Contract No. DE-0000206-1577) and by the National Science Foundation (Award No. EEC-0540879 to the Synthetic Biology Engineering Research Center, Award No. MCB-1341894, and Grant Nos. DGE 1106400 and 1106400 of the Graduate Research Fellowship Program).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This paper is dedicated to Professor Julian Davies, chemist, biochemist, microbiologist, and natural products ecologist, for his inspiration and leadership.
Rights and permissions
About this article

Cite this article
Yuzawa, S., Keasling, J. & Katz, L. Bio-based production of fuels and industrial chemicals by repurposing antibiotic-producing type I modular polyketide synthases: opportunities and challenges. J Antibiot 70, 378–385 (2017). https://doi.org/10.1038/ja.2016.136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2016.136
This article is cited by
-
Emerging evolutionary paradigms in antibiotic discovery
Journal of Industrial Microbiology and Biotechnology (2019)
-
Barriers and opportunities in bio-based production of hydrocarbons
Nature Energy (2018)
