
Abstract
A new xanthone named sydoxanthone C (1) and a new alkaloid named acremolin B (2), together with 10 known compounds (3–12) were isolated from a deep-sea-derived fungus Aspergillus sp. SCSIO Ind09F01. The structures of compounds (1–12) were determined by the extensive 1D, 2D-NMR, High resolution mass spectra (HRESIMS) data. Compounds 7, 8, 11 and 12 showed significant selective cytotoxicities against HeLa, DU145 and U937 cell lines. In addition, compounds 7, 8 and 11 also exhibited COX-2 inhibitory activities with the prominent IC50 values of 2.4, 7.1 and 10.6 μM, respectively.
Similar content being viewed by others
Introduction
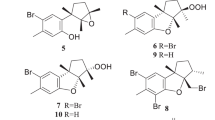
Marine-derived fungi tend to produce structurally unique and biologically active natural products, which were documented in recent years. Marine fungi, has provided more than 1500 new natural products from 2010 to 2014, with a diverse array of chemical moieties including polyketides, alkaloids, terpenoids, peptides, prenylated polyketides and lipids.1, 2, 3, 4 Especially, fungi from the deep-sea sediment and its derived compounds grabbed the attention of pharmaceutical chemists much in terms of its potential bioactivity. The deep sea is emerging as a new and interesting source of such microbes; however, only a handful of reports have described new metabolites from fungi derived from this habitat.5 As a wide stretch of this particular research theme, our present work resulted in the isolation of two new compounds along with 10 known compounds from the deep-sea-derived fungal strain Aspergillus sp. SCSIO Ind09F01 isolated from the deep-sea sediment sample of Indian Ocean. Our preliminary experiment demonstrated that crude extract of the culture of Aspergillus sp. SCSIO Ind09F01 showed toxicity toward the brine shrimp. Further solid-phase fermentation of Aspergillus sp. SCSIO Ind09F01 followed by its crude ethyl acetate (EtOAc) extract yielded two new compounds: sydoxanthone C (1) and acremolin B (2) together with 10 known compounds (3-12): sydoxanthone B (3),6 engyodontiumone B (4),7 sydowinin B (5),8 acremolin (6),9 diorcinol (7),10 cordyol C (8),11 cordyol E (9),12 methyl 2-hydroxy-4-(3-hydroxyl-5-methylphenoxy)-6-methylbenzoate(10),13 3,7-dihydroxy-1,9-dimethyldibenzofuran (11)14 and AGI-B4 (12)15 (Figure 1). The cytotoxicity and COX-2 inhibitory activities of these compounds were individually evaluated. Herein, we described the isolation, structure elucidation and bioactivities of these metabolites 1–12 from Aspergillus sp. SCSIO Ind09F01.

Structures of compounds 1–12.
Results and Discussion
Compound 1 was obtained as a yellow amorphous solid. The molecular formula of 1 was established as C17H14O7S by High resolution mass spectra (HRESIMS) (363.0521 [M+H]+), requiring 11 degrees of unsaturations. The 13C NMR, DEPT and HMQC spectra revealed the presence of 17 carbons, including 2 methyls (1 oxygenated), 1 oxygenated methylene, 4 aromatic methines, 8 aromatic quaternary carbons of which 3 were oxygenated and 2 carbonyl carbons. In the 1H NMR spectrum, the most salient signals were 2 methyls at δH 2.80 (3H, s), 3.95 (3H, s), a methylene at δH 4.61 (2H, s), four aromatic methines at δH 6.81 (1H, s), 7.05 (1H, s), 8.01 (1H, d, J=8.5 Hz), 8.39 (1H, d, J=8.5 Hz) (Table 1). Comparison of UV-vis and NMR data with those of sydoxanthone B (3)6 revealed a high degree of similarity. The only difference was the substituent of C-2, especially the chemical shift values of H3-12 and C-12. Meanwhile, the strong correlation from H3-12 (δH 2.80) to C-2 (δC 138.9) requirements of HRESIMS spectrum suggested that a methyl sulfonyl group was connected to C-2. Therefore, the structure of compound 1 was determined as pictured (Figure 2) and named sydoxanthone C.

Key HMBC correlations of compounds 1 and 2.
Compound 2 was obtained as a white amorphous solid and has a molecular formula of C12H15N5O as inferred from its HRESIMS (m/z 246.1346 [M+H] +) requiring eight degrees of unsaturations. The 1H NMR spectrum contained 15 proton signals: 4 sp3 methyls at δH 1.26 (6H, d, J=7 Hz), 3.54 (3H, s), 4.02 (3H, s), 1 sp3 aliphatic methine at δH (1H, sept, J=7 Hz), 2 sp2 methines at δH 7.58 (1H, s) and 7.82 (1H, s). The 13C NMR spectrum showed 11 carbon signals (2 overlapping carbon signals). There are six olefinic carbons (δC 103.7, 118.1, 133.7, 138.6, 142.9, 148.4) besides four methyls (δC 22.0, 22.0, 29.1, 32.7), one methine (δC 27.6) and one carbonyl carbon (155.1). Its NMR data are similar to that of acremolin (6)9 with the only difference in the presence of one extra methyl (δH 4.02 and δc 32.7), which suggests that compound 2 has the same skeleton as that of acremolin (6) with the only difference being the substitution at N-9. This was proved by the HMBC spectrum showing correlation of H3-11(δH 4.02) with C-8(δC 138.6)/C-4 (δC 133.7) (Table 2). Therefore, the structure of 2 was determined and projected as shown (Figure 2) and named acremolin B.
Compounds 1-12 were assessed for its cytotoxicity efficacy against K562, MCF-7, HeLa, DU145, U937, H1975, SGC-7901, A549, MOLT-4 and HL60 cell lines by standard MTT screening method. Trichostatin A and taxol are used as a positive control. Finally, Compounds 7, 8, 11 and 12 showed moderate selective cytotoxicity against HeLa, DU145 and U937 cell lines (Table 3). All the compounds were subjected for COX-2 inhibitory activity, where compounds 7, 8 and 11 also exhibited moderate COX-2 inhibitory activity with the IC50 values being of 2.4, 7.1 and 10.6 μM, respectively.
Experimental procedure
General experimental procedures
The NMR spectra were measured on a Bruker AC 500 MHz NMR (Bruker, Fällanden, Switzerland) spectrometer with TMS as an internal standard. HRESIMS were recorded on a Bruker micro TOF-QII mass spectrometer (Bruker, Fällanden, Switzerland). CD spectra were measured with a Chirascan circular dichroism spectrometer (Applied Photophysics, Surrey, UK). Size exclusion chromatography was done on Sephadex LH-20 gel (GE Healthcare, Uppsala, Sweden). Column chromatography was carried out on silica gel (200–300); Qingdao Marine Chemical Factory, Qingdao, China. Single-crystal data were measured on an Oxford Gemini S Ultra diffractometer (Agilent Technologies, Santa Clara, CA, USA). Spots were detected on TLC under UV light or by heating after spraying with 12% H2SO4 in H2O.
Fungal material
The fungal strain SCSIO Ind09F01 was isolated from the deep-sea sediment, which was collected from the Indian Ocean (Lat: 82.04513333' N, Long: 0.497883333' E) at the depth of 4530 m in 2013. The isolates were stored on MB agar (malt extract 15 g, sea salt 10 g, agar 15 g) slants at 4 °C and then deposited at CAS Key Laboratory of Tropical Marine Bio-resources and Ecology. The strain was identified to be a member of the Ascomycota generic on the basis of it’s ITS phylogenetic analysis, and it was named as Aspergillus sp. SCSIO Ind09F01. The 528 base-pair ITS sequence (NCBI GenBank accession number KP681643) has 99% sequence identity to that of Aspergillus sydowii strain NRRL 254 (NCBI GenBank accession number AY373869).
Fermentation, extraction and isolation
Aspergillus sp. SCSIO Ind09F01 was cultured on MB-agar plates at 25 °C for 7 days. The seed medium consisted of malt extract: 15 g, sea salt: 2.5 g, distilled water: 1000 ml, pH 7.4–7.8 was inoculated with strain SCSIO Ind09F01 and incubated at 25 °C for 72 h on a rotating shaker (170 r.p.m.). Mass scale fermentation of fungal isolate SCSIO Ind09F01 was carried out using production medium of solid rice in 1000 ml flasks (rice 200 g, sea salt 2.5 g, distilled water 200 ml) was inoculated with 10 ml of seed solution. Flasks were incubated at 25 °C under normal day night cycle. After 30 days, cultures from 30 flasks were harvested and subjected for organic extraction using EtOAc.
The EtOAc extracts of rice solid media of Aspergillus sp. SCSIO Ind09F01 were partitioned between petroleum ether, and 90% aqueous MeOH, The resulting MeOH phase was fractionated using silica column, Sephadex LH-20, and then semi-preparative reversed-phase (SP-RP) HPLC that yielded compounds 1–12 (Figure 1).
The culture of solid rice medium was soaked in acetone and cut into small pieces and kept for 1 day. The content was filtered and evaporated under vacuum using a Buchner funnel and extracted with EtOAc until exhaustion and this process was repeated thrice. The organic phase was collected and evaporated to obtain a dark brown oil crude extract (31.3 g). The crude EtOAc extract was subjected to silica gel column chromatography eluted with petroleum ether/EtOAc in a gradient eluent (v/v, 50:1, 30:1, 20:1, 10:1, 5:1, 1:1, 0:1) to obtain eight fractions (fractions 1–8) on the basis of TLC. Fraction 3 (4.1 g) was purified by Sephadex LH-20 (CHCl3/MeOH, 1:1) to afford compound 7 (3.0 g). Fr. 4 (400.5 mg) was purified by Sephadex LH-20 (CHCl3/MeOH, 1:1) to give three subfractions (fr. 4.1–4.3). Fr.4.2 (110.1 mg) was further purified by SP-RP HPLC using a C18 column (YMC-Pack, ODS-A S-5 μm × 12 nm 250 × 20 mm i.d., 4 ml per minute) eluting with MeOH/H2O (60:40) to afford compounds 8 (5.9 mg) and 11 (3.7 mg). Fr. 7 (3.5 g) was subjected to ODS chromatography eluted with MeOH/H2O in gradient eluent (1:9, 2:3, 3:2, 4;1, 9:1) to give three subfractions (fr. 7.1–7.3). Fr. 7.2 (900 mg) was purified by Sephadex LH-20 (MeOH) to give four subfractions (fr. 7.2.1–7.2.4). Fr. 7.2.2 (147 mg) was further purified by SP-RP HPLC eluting with CH3CN-H2O (50:50) to afford compounds 2 (35.6 mg) and 6 (8.9 mg). Fr. 8 (4.3 g) was subjected to silica gel column chromatography eluted with a CHCl3/MeOH in a step-wise gradient system at the ratios of 100:0, 50:1, 25:1, 10:1 and 0:100 (v/v) which yielded seven fractions. Fr. 8.2 (1.3 g) was subjected to ODS chromatography eluted with MeOH/H2O (linear gradient, 50% MeOH–100% MeOH) to give five subfractions (fr. 8.2.1–8.2.5). Fr. 8.2.3 (70.1 mg) was purified by SP-RP HPLC eluting by CH3CN- H2O (50:50) to afford compounds 1 (10.2 mg) and 4 (8.9 mg). Fr. 8.2.2 (4.7 mg) was purified by (SP-RP) HPLC eluting with CH3CN-H2O (50:50) to afford compounds 3 (3.3 mg) and 5 (8.0 mg). Fr. 8.2.5 (42.1 mg) was purified by (SP-RP) HPLC eluting with CH3CN-H2O (75:25) to afford compounds 12 (20.6 mg). Fr. 8.3 (1.1 g) was subjected to ODS chromatography eluted with MeOH/H2O in gradient eluent (1:9, 2:3, 3:2, 4;1, 9:1) to give three subfractions (fr. 8.3.1–8.3.3). Fr. 8.3.3 (132.5 mg) was purified by (SP-RP) HPLC eluting with MeOH-H2O (50:50) to afford compounds 9 (22.3 mg) and 10 (7.8 mg).
Sydoxanthone C (1): Yellow amorphous solid; UV (MeOH) λ max (log ɛ) 368 (3.10), 260 (4.20), 238 (4.09), 222 (4.01) nm; FT-IR: 3361, 1626, 1022 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 363.0521 [M+H]+ (calcd for C17H14O7S, 363.0533).
Acremolin B (2): White amorphous solid; UV (MeOH) λ max (log ɛ) 257 (4.12), 248 (4.11), 227 (4.44), 219 (4.41) nm; FT-IR: 1683, 1614, 1519 cm−1; 1H and 13C NMR data, see Table 2; HRESIMS m/z 246.1346 [M+H]+ (calcd for C12H15N5O, 246.1349).
Biological activities
Cytotoxicity assay
The cytotoxic activity of compounds 1-12 was screened against the growth panel of 10 tumor cell lines of (K562, MCF-7, HeLa, DU145, U937, H1975, SGC-7901, A549, MOLT-4 and HL60) according to Bergeron et al.16
COX-2 inhibitory activity assay
COX-2, as a well-established target, is an inducible enzyme, which expression is activated by cytokines, mitogens, endotoxin and tumor promoters. The anti-inflammatory and analgesic properties of traditional Non Steroidal Anti Inflammatory Drugs (NSAIDs) are primarily due to the inhibition of COX-2.17 Hence, the compounds isolated were tested for COX-2 inhibitory activity using the COX (ovine) inhibitor screening kit,18 according to the manufacturer’s instructions. The test compounds were dissolved in DMSO and the final concentration was set as 10 μM. The percentage inhibition has been calculated by comparison with control incubations.
Conclusion
Two new compounds sydoxanthone C (1) and acremolin B (2) along with 10 known compounds (3-12) were isolated from a deep-sea-derived fungi Aspergillus sp. SCSIO Ind09F01. Sydoxanthone C (1) is an analog of sydoxanthone B (3), the only difference is the methyl sulfonyl group substitution instead of methylthio group on C-2 position. Acremolin B (2) is a first analog of acremolin after its structure was revised recently.9 All the compounds were assessed for its cytotoxic ability and COX-2 inhibition efficiency. As a matter of fact among those 12 compounds, diphenyl etheric metabolites (7, 8 and 11) showed moderate selective cytotoxicity and COX-2 inhibitory activity in the screening test. Noteworthy, 3.0 g of compound 7 was obtained which will make it a practical and useful starting point for combinatorial synthesize of its analog to explore further research in structure–activity relationships.
References
Blunt, J.W., Copp, B. R., Keyzers, R. A., Munro, M. H. & Prinsep, M. R. Marine natural products. Nat. Prod. Rep. 31, 160–258 (2014).
Proksch, P., Putz, A., Ortlepp, S., Kjer, J. & Bayer, M. Bioactive natural products from marine sponges and fungal endophytes. Phytochem. Rev. 9, 475–489 (2010).
Rateb, M. E. & Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 28, 290–344 (2011).
Swathi, J., Narendra, K., Sowjanya, K. & Satya, A. K. Marine fungal metabolites as a rich source of bioactive compounds. Afr. J. Biochem. Res 7, 184–196 (2013).
Lin, X. et al. A new cytotoxic sesquiterpene quinone produced by Penicillium sp. F00120 isolated from a deep sea sediment sample. Mar. Drugs 10, 106–115 (2012).
Song, X. Q. et al. Xanthone derivatives from Aspergillus sydowii, an endophytic fungus from the liverwort Scapania ciliate S. Lac and their immunosuppressive activities. Phytochem. Lett 6, 318–321 (2013).
Yao, Q. et al. Cytotoxic polyketides from the deep-sea-derived fungus Engyodontium album DFFSCS021. Mar. Drugs 12, 5902–5915 (2014).
Hamasaki, T., Sato, Y. & Hatsuda, Y. Structure of sydowinin A, sydowinin B, and sydowinol, metabolites from Aspergillus sydowi.. Agri. Biol. Chem. 39, 2341–2345 (1975).
Januar, L. A. & Molinski, T. F. Acremolin from Acremonium strictum is N, 2, 3-Etheno-2′-isopropyl-1-methylguanine, not a 1 H-Azirine. Synthesis and Structural Revision. Org. Lett. 15, 2370–2373 (2013).
Yurchenko, A. N. et al. A new meroterpenoid from the marine fungus Aspergillus versicolor (Vuill.) Tirab. Russ. Chem. Bull. 59, 852–856 (2010).
Bunyapaiboonsri, T., Yoiprommarat, S., Intereya, K. & Kocharin, K. New diphenyl ethers from the insect pathogenic fungus Cordyceps sp. BCC 1861. Chem. Pharm. Bull. 55, 304–307 (2007).
Chen, M. et al. Bioactive indole alkaloids and phenyl Ether derivatives from a marine-derived Aspergillus sp. fungus. J. Nat. Prod. 76, 547–553 (2013).
Gong, D. L. et al. Diphenyl etheric metabolites from Streptomyces sp. neau50. J. Antibiot. 64, 465–467 (2011).
Takenaka, Y., Hamada, N. & Tanahashi, T. Monomeric and dimeric dibenzofurans from cultured mycobionts of Lecanora iseana. Phytochemistry 66, 665–668 (2005).
Kim, H. S. et al. A novel dihydroxan-thenone, AGI-B4 with inhibition of VEGF-induced endothelial cell growth. J. Antibiot. 55, 669–672 (2002).
Bergeron, J. J. M., Brenner, M. B., Thomas, D.Y. & Williams, D. B. Calnexin: a membrane bound chaperone of the endoplasmic reticulum. Trends Biochem. Sci. 19, 124–128 (1994).
Praveen Rao, P. N. & Edward, E. K. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): Cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharmaceut. Sci 11, 81–110 (2008).
Blobaum, A. L. et al. The 2′-Trifluoromethyl analogue of indomethacin is a potent and selective COX-2 inhibitor. ACS Med. Chem. Lett. 4, 486–490 (2013).
Acknowledgements
This work was supported by grants from the National Key Basic Research Program of China (973)’s Project (2011CB915503); the National High Technology Research and Development Program (863 Program, 2012AA092104), Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA11030403), the National Natural Science Foundation of China (31270402, 21172230, 41476135, 41376162, 41406187, 41406187); and Guangdong Marine Economic Development and Innovation of Regional Demonstration Project (GD2012-D01-001), Guangzhou Scientific Research Project (2014J4100212).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Tian, Y., Qin, X., Lin, X. et al. Sydoxanthone C and acremolin B produced by deep-sea-derived fungus Aspergillus sp. SCSIO Ind09F01. J Antibiot 68, 703–706 (2015). https://doi.org/10.1038/ja.2015.55
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2015.55
