
Abstract
The first total synthesis of ent-(+)-cinanthrenol A of potent estrogenic activity was achieved with 10.9% overall yield in 13 steps from commercially available materials. Our synthesis features a photo-promoted oxidative 6π-electron electrocyclization/aromatization for construction of the cyclopenta[a]phenanthren-17-one and Furukawa hydroxyl-directed cyclopropanation for the rare spiro[2,4]heptane. The brevity of this synthetic strategy would allow an expedited access to cinanthrenol A and its analogs for further biological evaluation.
Similar content being viewed by others

Introduction
Cinanthrenol A1 (Figure 1) was isolated in 2014 by Nakao and co-workers from the deep-water marine sponge Cinachyrella sp.2, 3, 4 and has shown moderate cytotoxicity against two cancer cell lines P-388 and HeLa with IC50 4.5 and 0.4 μg ml−1, respectively. Remarkably, cinanthrenol A was found to show estrogenic activity: binding strongly to estrogen receptor (ER) with IC50 value of 10 nM and changing the gene expression level of ER responsive genes A-MYB and SMAD3 in MCF-7 cells in a way similar to estradiol (E2).5 This estrogenic activity was believed to arise from its aromatic steroid structure that resembles estradiol and estrone (Figure 1). Structurally, cinanthrenol A contains a rare spiro[2,4]heptane and a cyclopenta[α]phenanthrene core, a common structural motif of widespread and carcinogenic steroid-related products (for example, 11-methyl-cyclco[α]phenanthren-17-one) found in petroleum, mineral oils, coal, lake sediments and cooking oils.6, 7 In addition, the presence of a hydroxyl group at C2, not C3, is very rare in the families of cyclopenta[α]phenanthrenes and steroids. The natural scarcity (3.5 mg obtained from 6.5 kg of the deep-water marine sponge, 5.4 × 10−5 wt% yield), potential biological activities, and the novel structure prompted us to undertake a synthetic study, culminating in the first total synthesis of cinanthrenol A.

Estradiol, estrone, and cinanthrenol A. A full color version of this figure is available at The Journal of Antibiotics journal online.
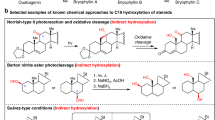
While many triaromatic steroids serving as useful geochemical biomarkers in fossil fuels are usually synthesized by multiple aromatizations of the corresponding steroids with low overall yields,8, 9, 10, 11, 12, 13 the de novo synthesis of cyclopenta[α]phenanthrenes has been underdeveloped in the past decades. Only three different approaches have been reported (Scheme 1a): (i) Friedel–Crafts acylative cyclization of tricyclic dihydrophenanthrene derivatives followed by oxidative aromatization by Coombs et al.,14, 15 (ii) acid-mediated alkylative cyclization of cyclopentanone-tethered naphthalene derivatives and oxidative aromatization by Lee and Harvey,16 and (iii) intermolecular Diels–Alder cycloaddition of Dane’s diene and α-bromocyclopentenone followed by dehydrogenative aromatization by Woski and Koreeda.17 Herein, we report a new strategy that revolves upon photo-promoted oxidative 6π-electron electrocyclization/aromatization to construct B-ring of the cyclopenta[α]phenanthren-17-one from readily available cis-stilbene-type compound 5, leading to the first total synthesis of cinanthrenol A.
Retrosynthetically, as depicted in Scheme 1b, we proposed Furukawa hydroxyl-directed cyclopropanation18 of 2 to install the unusual spiro[2,4]heptane. The allylic alcohol 2 could be prepared from ketone 3 through α-hydroxylation and Wittig olefination. Excellent Z/E selectivity was expected to avoid the potential steric interaction of methyl group and H12 of cyclopenta[α]phenanthrenone’s ortho-hydrogen. The cyclopenta[α]phenanthren-17-one 3 could be constructed by photo-promoted oxidative 6π-electron electrocyclization19 of cis-stilbene 5, which would be readily available from the double Sonogashira coupling of aryl iodides 6 and 7 with alkyne 8, followed by partial hydrogenation with Lindlar’s catalyst.
Results and Discussion
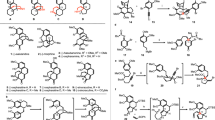
Our synthesis began with the preparation of alkyne 9 from the aryl iodide 6 (Scheme 2) through a high yielding three-step sequence: triisopropylsilyl (TIPS) protection, Sonogashira coupling, and chemoselective desilylation of trimethylsilyl (TMS) group. Alkyne 9 was subjected to another Sonogashira coupling with commercially available iodoindanone (7) under identical condition, providing alkyne 10 in a quantitative yield. After preliminary condition screenings of partial hydrogenation of the internal diarylalkyne, we quickly identified an optimized condition: 30 mol% Lindlar’s catalyst, 3 equivalents of quinoline, hydrogen gas (1 atm, balloon), and MeOH (solvent) at room temperature (RT) for 6 days, producing the desired cis-alkene 5a in excellent yield (85%). The oxidative 6π-electron electrocyclization proved to be challenging and extensive reaction conditions have been investigated for a better yield on a synthetically useful scale (Table 1). Initially, an oxidative thermal electrocyclization of 5a was examined under classical conditions (entries 1-3). Apparent decomposition was observed under all attempted conditions (FeCl320 at RT, DMSO/DMF21 at 140 °C, and ortho-dichlorobenenze at 200 °C) without any detectable cyclization product 3a. As well-recognized, photoreactions were wavelength-dependent. Therefore, a suitable wavelength for UV irradiation should be identified for I2-promoted oxidative photocyclization.22 When 180 nm wavelength was used (entry 4), significant decomposition was observed. Although irradiation with 300 nm gave only trace amount of 3a (mainly isomerization of cis-alkene to trans-alkene); 254 nm was found to be the optimal wavelength for the desired 6π electron electrocyclization, providing cyclopenta[α]phenanthren-17-one 3a in synthetically meaningful yield (20%). Further optimization of the reaction conditions including reaction time (entry 8 and 9), solvent23 (entry 10), oxidant (I2 vs PIDA,24 entry 13), and additive (propene oxide) (Katz’s condition,25, 26, 27, 28 entry 11) allowed us to increase the overall yield to be synthetically useful (50%). In addition, cyclopenta[α]phenanthren-17-ol derivative 5b was prepared from 5a to examine the key oxidative electrocyclization because the unfavorable influence of ketone group (c.f., 5a) on the oxidative photocyclization were on record.29, 30 However, no better yield (40% of 3b) was obtained from 5b under various electrocyclization conditions attempted for 5a. Finally, it should be noted that the concentration of the oxidative photocyclization must be within 0.001 M, because higher concentration resulted in considerable intermolecular [2+2] cycloaddition and decomposition (entry 7 in Table 1).
After several repeated operations of oxidative photocyclization to provide sufficient amount of cyclopenta[α]phenanthren-17-one 3a, we turned our attention to construct the unusual spiro[2,4]heptane through Furukawa modification of Simons–Smith cyclopropanation (Scheme 3). To this end, manipulations of ketone 3a were needed to prepare enantiomerically pure allylic alcohol 2 for hydroxyl-directed cyclopropanation. Although the classical Rubottom hydroxylation31 of the corresponding silyl enol ether proceeded smoothly to produce the desired α-hydroxyl ketone (±)-11 in 70% yield, an enantioselective α-hydroxylation of ketone 3a using Davis’ (S)-oxaziridine,32 surprisingly, gave (−)-11 with only 60% ee in 50% yield. The use of Davis’ (R)-oxaziridine led to the similar result: a 65% yield of with 50% ee. It was found that the dichloro derivative of Davis’ (R)-oxaziridine could deliver (+)-11 with a slight better ee value (75% ee) in 60% yield. In addition, we have made considerable attempts to increase the enantioenrichment of 11 including kinetic resolution via Sharpless asymmetric epoxidation protocol or lipase-mediated acetylation at a later stage without success.33 Therefore, we chose to move forward with (+)-11 (75% ee). It was noted that free alcohol of (+)-11 did not undergo efficient Wittig olefination. Upon temporary protection as TBS ether, Wittig olefination with triphenyl phosphonium ylide generated in situ occurred efficiently to provide alkene (−)-12 in 90% yield with excellent Z-selectivity (Z/E ⩾10:1), which was confirmed by nOe experiments. As chemoselective desilylation (TBS over TIPS) was problematic, desilylation of both TIPS and TBS groups with TBAF and regioselective acetylation of the aromatic alcohol were performed to provide the allylic alcohol (+)-2. It should be noted that the free phenol caused significant decomposition in the subsequent cyclopropanation. The Furukawa–Simons–Smith18 cyclopropanation of (+)-2 proceeded smoothly with Et2Zn and diiodomethane to furnish ent-(+)-cinanthrenol A (ent-1) in 78% yield after deacetylation with K2CO3 in methanol. Remarkably, a single diastereomer was observed in cyclopropanation reaction, in supportive of the high hydroxyl-directed diastereoselectivity.34 The opposite sign of optical rotation of synthetic ent-1 (derived from (+)-11 in 75% ee) and natural cinanthrenol A provided a confirmation of its absolute configuration ([a]D=+6.3, c 0.3, MeOH; lit[a]D=−11.6, c 0.16, MeOH). However, the ee% value of our synthetic cinanthrenol A (ent-1) was not clear.
In summary, we have accomplished the first total synthesis of ent-(+)-cinanthrenol A with 10.9% overall yield in 13 steps from commercially available starting materials. Our synthesis was enabled by some key reactions including Sonogashira coupling, oxidative 6π-electron electrocyclization, Davis’ asymmetric hydroxylation, and Furukawa–Simons–Smith cyclopropanation. Our synthetic studies confirmed the absolute configuration of natural cinanthrenol A and provide an expedient access to the analogs for further biological evaluation.
Materials and methods
Reactions were carried out in oven or flame-dried glassware under a nitrogen atmosphere, unless otherwise noted. Tetrahydrofuran (THF) was freshly distilled before use from sodium using benzophenone as indicator. Dichloromethane was freshly distilled before use from calcium hydride (CaH2). All other solvents were dried over 3 or 4 Å molecular sieves. Solvents used in workup, extraction, and column chromatography were used as received from commercial suppliers without prior purification. Reactions were magnetically stirred and monitored by thin layer chromatography (TLC, 0.25 mm) on Merck pre-coated silica gel plates (Sigma-Aldrich, St Louis, MO, USA). Flash chromatography was performed with silica gel 60 (particle size 0.040–0.062 mm) supplied by Grace. IR spectra were collected on a Bruker model TENSOR27 spectrophotometer (Bruker, Billerica, MA, USA). 1H and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer (400 MHz for 1H, 100 MHz for 13C) (Bruker). Chemical shifts are reported in p.p.m. as values relative to the internal chloroform (7.26 p.p.m. for 1H and 77.0 p.p.m. for 13C) or pyridine (7.55 p.p.m. (tr) for 1H and 135.5 p.p.m. (tr) for 13C). Abbreviations for signal coupling are as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Optical rotations were measured on a JASCO Perkin–Elmer model P-2000 polarimeter (JASCO, Easton, MD, USA). High-resolution mass spectra were measured at the Hong Kong University of Science and Technology Mass Spectrometry Service Center on either an Agilent GC/MS 5975C system (Agilent, Santa Clara, CA, USA) or an API QSTAR XL System (Applied Biosystems, Waltham, MA, USA).

(a) Previous synthetic strategies for cyclopenta[α]phenanthren-17-one and (b) retrosynthetic analysis of cinanthrenol A. A full color version of this scheme is available at The Journal of Antibiotics journal online.

Construction of cyclopenta[α]phenanthren-17-one 3a. A full color version of this figure is available at The Journal of Antibiotics journal online.

Total Synthesis of ent-(+)-Cinanthrenol A. A full color version of this figure is available at The Journal of Antibiotics journal online.
Preparation of alkyne 9
To a stirred solution of 4-iodophenol 6 (2.20 g, 10 mmol) in DMF (20 ml) at 0 °C were added imidazole (1.70 g, 25.0 mmol) and triisopropylsilyl chloride (2.67 mL, 12.5 mmol). The reaction mixture was allowed to warm to RT and stirred overnight. The reaction was quenched by addition of aq. satd. NH4Cl (50 ml). The organic layer was collected and the aqueous layer was extracted with Et2O (3 × 50 ml). The combined organic fractions were washed with H2O (3 × 30 ml), brine, dried over MgSO4, and concentrated under reduced pressure to give the crude TIPS ether S-1. A small amount of the residue was purified by flash column chromatography on silica gel using eluents (ethyl acetate/hexane=1/10–1/5) to give the pure S-1 as a colorless oil for data collection. IR (neat, cm−1): 2945, 2892, 2867, 1581, 1485, 1388, 1273, 1168, 1003, 909, 883, 825, 726, 686. 1H NMR (400 MHz, CDCl3) δ 7.49 (d, J=8.4 Hz, 2H), 6.65 (d, J=8.4 Hz, 2H), 1.33–1.16 (m, 3H), 1.09 (d, J=7.2 Hz, 18H). 13C NMR (100 MHz, CDCl3) δ 156.0, 138.2 (2C), 122.3 (2C), 83.2, 17.9 (6C), 12.6 (3C). HRMS (CI+): m/z calculated for C15H25IOSi [M]+ 376.0719, found 376.0712.
To a stirred solution of crude S-1 in dry THF (50 ml) at RT were added bis(triphenylphosphine)palladium(II) dichloride (351 mg, 0.5 mmol), CuI (195 mg, 1.0 mmol), triethylamine (4.18 ml, 30.0 mmol) and trimethylsilylacetylene 8 (7.0 ml, 50.0 mmol). The reaction mixture was refluxed overnight, cooled to RT, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (ethyl acetate/hexane=1/10–1/5) to give the S-2 as a colorless oil. IR (neat, cm−1): 2947, 2895, 2869, 2158, 1602, 1505, 1278, 1250, 1096, 910, 865, 840, 686. 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J=8.4 Hz, 2H), 6.79 (d, J=8.4 Hz, 2H), 1.30–1.16 (m, 3H), 1.08 (d, J=7.2 Hz, 18H), 0.23 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 156.5, 133.4 (2C), 119.9 (2C), 115.5, 105.3, 92.5, 17.8 (6C), 12.6 (3C), 0.0 (3C). HRMS (CI+): m/z calculated for C20H34OSi2 [M]+ 246.2148, found 246.2155.
To a stirred solution of the alkyne S-2 in MeOH (25 ml) and CH2Cl2 (25 ml) at RT was added potassium carbonate (138 mg, 1.0 mmol). The reaction mixture was stirred overnight at RT, filtered through a short pad of Celite. The filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (EtOAc/hexane=1/20) to give the alkyne 9 (2.47 g, 90% over 3 steps) as a colorless oil. IR (neat, cm−1): 2946, 2893, 2868, 1603, 1505, 1464, 1278, 910, 882, 839, 686. 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J=8.4 Hz, 2H), 6.82 (d, J=8.4 Hz, 2H), 3.00 (s, 1H), 1.31–1.18 (m, 3H), 1.09 (d, J=7.6 Hz, 18H). 13C NMR (100 MHz, CDCl3) δ 156.7, 133.6 (2C), 119.9 (2C), 114.4, 83.8, 75.8, 17.8 (6C), 12.6 (3C). HRMS (CI+): m/z calculated for C17H27OSi [M+H]+ 275.1831, found 275.1844.
Preparation of diarylalkyne 10
To a stirred solution of the alkyne 9 (2.47 g, 9.0 mmol) and 4-iodoindanone 7 (1.55 g, 6.0 mmol) in dry THF (50 ml) at RT were added bis(triphenylphosphine)palladium(II) dichloride (211 mg, 0.3 mmol), CuI (117 mg, 0.6 mmol) and triethylamine (2.53 ml, 18.0 mmol). The reaction mixture was refluxed overnight, cooled to RT, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (ethyl acetate/hexane=1/10–1/5) to give the diarylalkyne 10 (2.41 g, >99% yield) as a brown oil. IR (neat, cm−1): 3039, 2945, 2892, 2867, 1719, 1597, 1509, 1463, 1276, 908, 883, 839, 778, 687. 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J=7.6 Hz, 2H), 7.42 (d, J=8.4 Hz, 2H), 7.33 (t, J=7.6 Hz, 1H), 6.87 (d, J=8.4 Hz, 2H), 3.30–3.13 (m, 2H), 2.78–2.65 (m, 2H), 1.36–1.19 (m, 3H), 1.10 (d, J=7.6 Hz, 18H). 13C NMR (100 MHz, CDCl3) δ 206.6, 156.9, 156.7, 137.1, 136.5, 133.0 (2C), 127.4, 122.9, 122.5, 120.0 (2C), 115.0, 95.0, 84.3, 36.0, 25.5, 17.8 (6C), 12.6 (3C). HRMS (CI+): m/z calculated for C26H33O2Si [M+H]+ 405.2250, found 405.2263.
Preparation of cis-stilbene 5a
To a stirred solution of the diarylalkyne 10 (2.41 g, 6.0 mmol) in MeOH (30 ml) at RT were added Lindlar’s catalyst (3.82 g, 1.8 mmol), quinoline (2.13 ml, 18.0 mmol). The reaction mixture was flushed with hydrogen gas three times. The reaction mixture under H2 atmosphere (1.0 atm, balloon) was stirred at RT for 6 days. The reaction mixture was filtered through a pad of Celite and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (ethyl acetate /hexane=1/10–1/5) to give the cis-stilbene 5a (2.07 g, 85% yield) as a brown oil. IR (neat, cm−1): 2944, 2867, 1716, 1601, 1509, 1464, 1270, 1169, 1090, 910, 883, 782, 686. 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J=7.6 Hz, 1H), 7.49 (d, J=7.6 Hz, 1H), 7.25 (t, J=7.6 Hz, 1H), 7.00 (d, J=8.4 Hz, 2H), 6.69 (m, 3H), 6.51 (d, J=12.0 Hz, 1H), 2.93–2.83 (m, 2H), 2.63–2.60 (m, 2H), 1.27–1.16 (m, 3H), 1.07 (d, J=7.2 Hz, 18H). 13C NMR (100 MHz, CDCl3) δ 207.3, 155.6, 153.7, 137.3, 136.4, 134.4, 132.1, 129.8 (2C), 129.6, 127.3, 124.3, 122.3, 119.9 (2C), 36.1, 25.0, 17.8 (6C), 12.6 (3C). HRMS (CI+): m/z calculated for C26H34O2Si [M]+ 406.2328, found 406.2328.
Preparation of cis-Stilbene 5b
To a stirred solution of ketone 5a (406 mg, 1.0 mmol) in MeOH (10 ml) was added sodium borohydride (56.7 mg, 1.5 mmol) at 0 °C. The reaction was stirred at 0 °C for 2 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (EtOAc/hexane=1/10–1/5) to give the secondary alcohol product. The alcohol product was dissolved in dry DMF (5 ml), and to the solution were added imidazole (170 mg, 2.5 mmol) and triethylsilyl chloride (0.20 ml, 1.2 mmol) at 0 °C. The reaction mixture was allowed to warm to RT and stirred overnight. The reaction was quenched by addition of aq. satd. NH4Cl (10 ml). The organic layer was collected and the aqueous layer was extracted with Et2O (3 × 20 ml). The combined organic fractions were washed with H2O (3 × 10 ml), brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (ethyl acetate/hexane=1/20–1/15) to give the cis-stilbene 5b (571 mg, 90% over 2 steps) as a brown oil. IR (neat, cm−1): 2948, 2869, 1603, 1508, 1462, 1267, 1106, 1013, 912, 883, 787, 744. 1H NMR (400 MHz, CDCl3) δ 7.22 (d, J=6.4 Hz, 1H), 7.11 (m, 2H), 7.04 (d, J=8.4 Hz, 2H), 6.70 (d, J=8.4 Hz, 2H), 6.51 (dd, J=32.2, 12.0 Hz, 2H), 5.22 (t, J=7.2 Hz, 1H), 2.82 (dd, J=15.6, 9.0 Hz, 1H), 2.51–2.40 (m, 1H), 2.40–2.30 (m, 1H), 1.84 (dq, J=12.4, 8.6 Hz, 1H), 1.28–1.17 (m, 3H), 1.08 (d, J=7.2 Hz, 17H), 1.03 (t, J=8.0 Hz, 10H), 0.71 (q, J=8.0 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 155.2, 145.9, 141.0, 134.3, 130.3, 130.3, 129.9 (2C), 127.7, 126.6, 126.5, 122.6, 119.7 (2C), 76.4, 36.3, 28.6, 17.9 (6C), 12.6 (3C), 6.9 (3C), 5.0 (3C). HRMS (CI+): m/z calculated for C30H50O2Si2 [M]+ 522.3349, found 522.3359.
Preparation of 3a/3b
To a round bottom quartz flask (150 ml) were added cis-stilbene 5a/5b (0.1 mmol), cyclohexane (100 ml), I2 (25 mg, 0.1 mmol), and propene oxide (1.16g, 1.4 ml, 20 mmol). The flask containing the reaction mixture was placed in a Rayonet chamber reactor (Rayonet-200 model, The Southern New England Ultraviolet Company, Branford, CT, USA) and irradiated with RPR-2537 lamps (λ=254 nm) at RT for 3–24 h. The reaction was quenched by addition of aq. satd. NaHCO3 (30 ml) and aq. satd. Na2S2O3 (30 ml). The organic layer was collected and the aqueous layer was extracted with Et2O (3 × 50 ml). The combined organic fractions were washed with brine, dried over MgSO4, and concentrated under reduced pressure to give the crude mixture, which was purified by flash column chromatography on silica gel. 3a was obtained in 50% yield (entry 11). IR (neat, cm−1): 2926, 2866, 1711, 1614, 1509, 1457, 1264, 1232, 991, 924, 882, 864, 670. 1H NMR (400 MHz, CDCl3) δ 8.49 (d, J=8.8 Hz, 1H), 8.11 (s, 1H), 7.88 (d, J=8.8 Hz, 1H), 7.83–7.76 (m, 2H), 7.71 (d, J=8.8 Hz, 1H), 7.27 (d, J=8.8 Hz, 1H), 3.41 (br s, 2H), 2.82 (br s, 2H), 1.36 (m, 3H), 1.16 (d, J=7.2 Hz, 18H). 13C NMR (100 MHz, CDCl3) δ 206.9, 155.4, 155.37, 134.9, 133.4, 131.7, 130.2, 129.2, 128.1, 127.9, 122.8, 122.4, 119.9, 119.4, 112.3, 36.3, 24.6, 18.0 (6C), 12.7 (3C). HRMS (CI+): m/z calculated for C26H32O2Si [M]+ 404.2172, found 404.2176.
3b was obtained in 40% yield from 5b. IR (neat, cm−1): 2947, 2869, 1617, 1600, 1508, 1457, 1232, 1103, 879, 834, 753, 685. 1H NMR (400 MHz, CDCl3) δ 8.46 (d, J=8.8 Hz, 1H), 8.11 (s, 1H), 7.76 (d, J=8.8 Hz, 1H), 7.70 (d, J=8.8 Hz, 1H), 7.62 (d, J=9.2 Hz, 2H), 7.19 (d, J=8.4 Hz, 1H), 5.49 (t, J=6.4 Hz, 1H), 3.45 (m, 1H), 3.09 (dt, J=16.0, 8.0 Hz, 1H), 2.65 (m, J=12.0, 8.0 Hz, 1H), 2.12 (m, 1H), 1.36 (m, 3H), 1.17 (d, J=7.6 Hz, 18H), 1.07 (t, J=8.0 Hz, 9H), 0.75 (q, J=8.0 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 154.9, 143.2, 139.6, 132.2, 129.9, 129.4, 128.8, 126.7 (2C), 122.3, 121.9, 120.9, 120.7, 111.8, 77.0, 36.2, 28.4, 18.0 (6C), 12.8 (3C), 6.9 (3C), 5.1 (3C). HRMS (CI+): m/z calculated for C32H48O2Si2 [M]+ 520.3193, found 520.3198.
Preparation of α-Hydroxyl Ketone 11
α-Hydroxylation: To a stirred solution of ketone 3a (202 mg, 0.5 mmol) in CH2Cl2 (5 ml) at 0 °C were added triethylamine (0.1 ml, 0.75 mmol) and tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf, 0.14 ml, 0.6 mmol). The reaction mixture was allowed to warm to RT and stirred for 2 h. The reaction was quenched by addition of aq. satd. NaHCO3 (10 ml). The organic layer was collected and the aqueous layer was extracted with CH2Cl2 (3 × 10 ml). The combined organic fractions were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The crude product was dissolved in CH2Cl2 (5 ml) at 0 °C, to the resulting solution were added NaHCO3 (84 mg, 1.0 mmol) and 3-chloroperbenzoic acid (168 mg, 0.75 mmol). The reaction mixture was allowed to warm to RT and stirred for 4 h. The reaction was quenched by addition of aq. satd. NaHCO3 (10 ml). The organic layer was collected and the aqueous layer was extracted with CH2Cl2 (3 × 10 ml). The combined organic fractions were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (ethyl acetate/hexane=1/6–1/3) to give the α-hydroxyl ketone 11 (147 mg, 70% yield) as a brown solid. Asymmetric α-Hydroxylation: to a stirred solution of sodium bis(trimethylsilyl)amide (1M in THF, 0.6 ml, 0.6 mmol) in THF (5 ml) at −78 °C was added the solution of ketone 3a (202 mg, 0.5 mmol) in THF (2 ml), the reaction mixture was stirred at −78 °C for 30 min. Then a solution of Davis’ (S)-oxaziridine (0.75 mmol) in THF (2 ml) was added via syringe, the reaction mixture was stirred at −78 °C for 30 min. The reaction was quenched by addition of aq. satd. NH4I (10 ml). The organic layer was collected and the aqueous layer was extracted with Et2O (3 × 10 ml). The combined organic fractions were washed with brine, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (ethyl acetate/hexane=1/6–1/3) to give the α-hydroxyl ketone 11 as a brown solid. (+)-11 was obtained following the same procedure using Davis’ (R)-oxaziridine or dichloride derivative of Davis’ (R)-oxaziridine. (+)-11 (75% ee):  = +7.5(c 1.0, CHCl3). IR (neat, cm−1): 3419, 2944, 2866, 1712, 1613, 1513, 1457, 1266, 1234, 983, 882, 841. 1H NMR (400 MHz, CDCl3) δ 8.57 (d, J=8.8 Hz, 1H), 8.13 (d, J=2.0 Hz, 1H), 7.91 (d, J=8.4 Hz, 1H), 7.85 (m, 2H), 7.76 (d, J=8.8 Hz, 1H), 7.31 (dd, J=8.8, 2.4 Hz, 1H), 4.72 (dd, J=7.6, 4.0 Hz, 1H), 4.03 (dd, J=16.8, 7.6 Hz, 1H), 3.27 (dd, J=16.8, 4.0 Hz, 1H), 1.38 (m, 3H), 1.17 (d, J=7.2 Hz, 18H). 13C NMR (100 MHz, CDCl3) δ 206.2, 155.6, 151.6, 134.4, 131.8, 131.7, 130.3, 129.1, 128.3, 128.3, 123.5, 122.7, 120.2, 119.4, 112.3, 74.2, 33.7, 18.0 (6C), 12.8 (3C). HRMS (CI+): m/z calculated for C26H32O3Si [M]+ 420.2121, found 420.2122.
= +7.5(c 1.0, CHCl3). IR (neat, cm−1): 3419, 2944, 2866, 1712, 1613, 1513, 1457, 1266, 1234, 983, 882, 841. 1H NMR (400 MHz, CDCl3) δ 8.57 (d, J=8.8 Hz, 1H), 8.13 (d, J=2.0 Hz, 1H), 7.91 (d, J=8.4 Hz, 1H), 7.85 (m, 2H), 7.76 (d, J=8.8 Hz, 1H), 7.31 (dd, J=8.8, 2.4 Hz, 1H), 4.72 (dd, J=7.6, 4.0 Hz, 1H), 4.03 (dd, J=16.8, 7.6 Hz, 1H), 3.27 (dd, J=16.8, 4.0 Hz, 1H), 1.38 (m, 3H), 1.17 (d, J=7.2 Hz, 18H). 13C NMR (100 MHz, CDCl3) δ 206.2, 155.6, 151.6, 134.4, 131.8, 131.7, 130.3, 129.1, 128.3, 128.3, 123.5, 122.7, 120.2, 119.4, 112.3, 74.2, 33.7, 18.0 (6C), 12.8 (3C). HRMS (CI+): m/z calculated for C26H32O3Si [M]+ 420.2121, found 420.2122.
Preparation of Alkene (−)-12
To a stirred solution of α-hydroxyl ketone (+)-11 (126 mg, 0.3 mmol, 75% ee) in DMF (5 ml) at 0 °C were added imidazole (51 mg, 0.75 mmol) and tert-butyldimethylsilyl chloride (54 mg, 0.36 mmol). The reaction mixture was allowed to warm to RT and stirred overnight. The reaction was quenched by addition of aq. satd. NH4Cl (10 ml). The organic layer was collected and the aqueous layer was extracted with Et2O (3 × 10 ml). The combined organic fractions were washed with H2O (3 × 10 ml), brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (ethyl acetate/hexane=1/20–1/15) to give the pure TBS ether S-3 (152 mg, 95%) as a colorless oil.  = −5.0 (c 0.5, CHCl3). IR (neat, cm−1): 2928, 2860, 1724, 1613, 1513, 1460, 1262, 1128, 868, 837, 672. 1H NMR (400 MHz, CDCl3) δ 8.54 (d, J=8.6 Hz, 1H), 8.13 (s, 1H), 7.90 (d, J=8.6 Hz, 1H), 7.84 (d, J=8.6 Hz, 2H), 7.75 (d, J=8.8 Hz, 1H), 7.30 (d, J=8.6 Hz, 1H), 4.77–4.64 (m, 1H), 3.95 (dd, J=16.6, 7.4 Hz, 1H), 3.24 (dd, J=16.6, 3.2 Hz, 1H), 1.43–1.33 (m, 3H), 1.17 (d, J=7.2 Hz, 18H), 1.00 (s, 9H), 0.27 (d, J=4.4 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 204.7, 155.5, 150.7, 134.0, 132.4, 131.8, 130.2, 129.0, 128.2, 128.0, 123.2, 122.6, 120.3, 119.5, 112.3, 74.8, 35.2, 25.9 (3C), 18.5, 18.0 (6C), 12.8 (3C), −4.3, −5.0. HRMS (CI+): m/z calculated for C32H47O3Si2 [M]+ 535.3064, found 535.3047.
= −5.0 (c 0.5, CHCl3). IR (neat, cm−1): 2928, 2860, 1724, 1613, 1513, 1460, 1262, 1128, 868, 837, 672. 1H NMR (400 MHz, CDCl3) δ 8.54 (d, J=8.6 Hz, 1H), 8.13 (s, 1H), 7.90 (d, J=8.6 Hz, 1H), 7.84 (d, J=8.6 Hz, 2H), 7.75 (d, J=8.8 Hz, 1H), 7.30 (d, J=8.6 Hz, 1H), 4.77–4.64 (m, 1H), 3.95 (dd, J=16.6, 7.4 Hz, 1H), 3.24 (dd, J=16.6, 3.2 Hz, 1H), 1.43–1.33 (m, 3H), 1.17 (d, J=7.2 Hz, 18H), 1.00 (s, 9H), 0.27 (d, J=4.4 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 204.7, 155.5, 150.7, 134.0, 132.4, 131.8, 130.2, 129.0, 128.2, 128.0, 123.2, 122.6, 120.3, 119.5, 112.3, 74.8, 35.2, 25.9 (3C), 18.5, 18.0 (6C), 12.8 (3C), −4.3, −5.0. HRMS (CI+): m/z calculated for C32H47O3Si2 [M]+ 535.3064, found 535.3047.
To a stirred suspension of C2H5PPh3Br (223 mg, 0.6 mmol) in THF (5 ml) at 0 °C was added sodium bis(trimethylsilyl)amide (1.0 M in THF, 0.5 ml, 0.5 mmol), the reaction mixture was stirred at 0 °C for 30 min. Then a solution of TBS ether S-3 (134 mg, 0.25 mmol) in THF (3 ml) was added via syringe, the reaction mixture was allowed to warm to RT and stirred overnight. The reaction was quenched by addition of aq. satd. NH4Cl (10 mL). The organic layer was collected and the aqueous layer was extracted with Et2O (3 × 10 ml). The combined organic fractions were washed with brine, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (ethyl acetate/hexane=1/20–1/15) to give the alkene (−)-12 (123 mg, 90% yield) as a brown oil.  = −8.0(c 0.1, CHCl3). IR (neat, cm−1): 2924, 2854, 1617, 1460, 1376, 1261, 1085, 1019, 800. 1H NMR (400 MHz, CDCl3) δ 8.39 (d, J=8.4 Hz, 1H), 8.07 (s, 1H), 7.75 (d, J=8.4 Hz, 1H), 7.70 (d, J=8.8 Hz, 1H), 7.65 (d, J=8.4 Hz, 1H), 7.57 (d, J=8.8 Hz, 1H), 7.18 (d, J=8.4 Hz, 1H), 6.30 (d, J=6.8 Hz, 1H), 5.38 (d, J=6.0 Hz, 1H), 3.62 (dd, J=16.8, 7.0 Hz, 1H), 3.20 (d, J=16.8 Hz, 1H), 2.02 (d, J=6.8 Hz, 3H), 1.37 (m, 3H), 1.17 (d, J=7.2 Hz, 18H), 0.93 (s, 9H), 0.18 (d, J=9.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 155.0, 145.2, 138.8, 138.1, 132.3, 129.9, 129.09, 129.06, 126.9, 126.7, 122.1, 120.72, 120.69, 119.2, 118.3, 111.6, 72.1, 40.5, 29.7, 25.9 (3C), 18.0 (6C), 14.7, 12.8 (3C), −3.7, −4.5. HRMS (CI+): m/z calculated for C34H50O2Si2 [M]+ 546.3349, found 546.3376.
= −8.0(c 0.1, CHCl3). IR (neat, cm−1): 2924, 2854, 1617, 1460, 1376, 1261, 1085, 1019, 800. 1H NMR (400 MHz, CDCl3) δ 8.39 (d, J=8.4 Hz, 1H), 8.07 (s, 1H), 7.75 (d, J=8.4 Hz, 1H), 7.70 (d, J=8.8 Hz, 1H), 7.65 (d, J=8.4 Hz, 1H), 7.57 (d, J=8.8 Hz, 1H), 7.18 (d, J=8.4 Hz, 1H), 6.30 (d, J=6.8 Hz, 1H), 5.38 (d, J=6.0 Hz, 1H), 3.62 (dd, J=16.8, 7.0 Hz, 1H), 3.20 (d, J=16.8 Hz, 1H), 2.02 (d, J=6.8 Hz, 3H), 1.37 (m, 3H), 1.17 (d, J=7.2 Hz, 18H), 0.93 (s, 9H), 0.18 (d, J=9.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 155.0, 145.2, 138.8, 138.1, 132.3, 129.9, 129.09, 129.06, 126.9, 126.7, 122.1, 120.72, 120.69, 119.2, 118.3, 111.6, 72.1, 40.5, 29.7, 25.9 (3C), 18.0 (6C), 14.7, 12.8 (3C), −3.7, −4.5. HRMS (CI+): m/z calculated for C34H50O2Si2 [M]+ 546.3349, found 546.3376.
Preparation of allylic alcohol (+)-2
To a solution of the allylic alcohol (−)-12 (109 mg, 0.2 mmol) in THF (3 ml) at 0 °C was added tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 0.6 ml, 0.6 mmol). The reaction mixture was allowed to warm to RT and stirred for 3 h. The reaction was quenched by addition of aq. satd. NH4Cl (10 ml). The organic layer was collected and the aqueous layer was extracted with EtOAc (3 × 10 ml). The combined organic fractions were washed with brine, dried over MgSO4, and concentrated under reduced pressure to give a crude product, which was washed through a short pad of silica gel using eluents (ethyl acetate) to remove TBAF to give the phenol intermediate (44 mg, 80%) as a yellow solid. To a stirred solution of the phenol intermediate (28 mg, 0.1 mmol) in CH2Cl2 (1 ml) at RT were added triethylamine (28 μl, 0.2 mmol) and acetic anhydride (14 μl, 0.15 mmol). The reaction mixture was stirred at RT for 2 h. The reaction was quenched by addition of aq. satd. NaHCO3 (5 ml). The organic layer was collected and the aqueous layer was extracted with CH2Cl2 (3 × 5 ml). The combined organic fractions were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (ethyl acetate/hexane=1/3–1/1) to give acetate (+)-2 (29 mg, 90%) as a colorless oil.  = −5.0(c 0.5, CHCl3). IR (neat, cm−1): 3420, 2924, 1755, 1619, 1370, 1213, 1165, 840, 755. 1H NMR (400 MHz, C6D6) δ 8.45 (d, J=8.4 Hz, 1H), 8.33 (s, 1H), 7.90 (d, J=8.8 Hz, 1H), 7.82–7.64 (m, 3H), 7.33 (d, J=8.4 Hz, 1H), 6.36 (d, J=7.2 Hz, 1H), 5.35–5.24 (m, 1H), 3.65 (dd, J=17.6, 6.2 Hz, 1H), 3.32 (d, J=17.6 Hz, 1H), 2.41 (s, 3H), 2.09 (d, J=6.4 Hz, 3H). 13C NMR (100 MHz, C6D6) δ 69.7, 149.4, 145.7, 139.2, 137.8, 131.7, 130.0, 129.8, 129.6, 129.3, 126.9, 123.0, 122.5, 120.8, 120.3, 118.8, 114.9, 71.1, 39.9, 21.3, 14.6. HRMS (CI+): m/z calculated for C21H18O3 [M]+ 318.1256, found 318.1262.
= −5.0(c 0.5, CHCl3). IR (neat, cm−1): 3420, 2924, 1755, 1619, 1370, 1213, 1165, 840, 755. 1H NMR (400 MHz, C6D6) δ 8.45 (d, J=8.4 Hz, 1H), 8.33 (s, 1H), 7.90 (d, J=8.8 Hz, 1H), 7.82–7.64 (m, 3H), 7.33 (d, J=8.4 Hz, 1H), 6.36 (d, J=7.2 Hz, 1H), 5.35–5.24 (m, 1H), 3.65 (dd, J=17.6, 6.2 Hz, 1H), 3.32 (d, J=17.6 Hz, 1H), 2.41 (s, 3H), 2.09 (d, J=6.4 Hz, 3H). 13C NMR (100 MHz, C6D6) δ 69.7, 149.4, 145.7, 139.2, 137.8, 131.7, 130.0, 129.8, 129.6, 129.3, 126.9, 123.0, 122.5, 120.8, 120.3, 118.8, 114.9, 71.1, 39.9, 21.3, 14.6. HRMS (CI+): m/z calculated for C21H18O3 [M]+ 318.1256, found 318.1262.
Preparation of ent-cinanthrenol A
To a stirred solution of (+)-2 (16 mg, 0.05 mmol) in CH2Cl2 (1 ml) at 0 °C was added diiodomethane (40 μl, 0.5 mmol) and diethylzinc solution (1M in hexanes, 0.5 ml, 0.5 mmol). The reaction mixture was stirred at 0 °C for 4 h. The reaction was quenched by addition of aq. satd. NaHCO3 (5 ml). The organic layer was collected and the aqueous layer was extracted with CH2Cl2 (3 × 5 ml). The combined organic fractions were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (ethyl acetate/hexane=1/3–1/1) to give the cyclopropane intermediate as a yellow solid. To a stirred solution of the cyclopropane derivative in MeOH (1 ml) at RT was added potassium carbonate (1.4 mg, 0.01 mmol). The reaction mixture was stirred at RT for 2 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel using eluents (ethyl acetate/hexane=1/3–1/1) to give the final product ent-1 (11 mg, 78% over 2 steps) as a yellow solid.  = +6.3(c 0.3, MeOH). IR (neat, cm−1): 3422, 2921, 2864, 1742, 1513, 1460, 1385. 1H NMR (400 MHz, Pyr) δ 12.00 (s, 1H), 8.64 (d, J=8.0 Hz, 1H), 8.57 (s, 1H), 7.94 (d, J=8.8 Hz, 1H), 7.84 (d, J=8.8 Hz, 1H), 7.64 (d, J=8.8 Hz, 1H), 7.52 (d, J=8.8 Hz, 1H), 6.98 (d, J=8.4 Hz, 1H), 6.51 (d, J=6.4 Hz, 1H), 4.86 (m, 1H), 3.87 (dd, J=16.8, 7.4 Hz, 1H), 3.61 (d, J=16.8 Hz, 1H), 1.46 (m, 1H), 1.36 (m, 1H), 1.30 (d, J=4.8 Hz, 3H), 1.16 (m, 1H). 13C NMR (100 MHz, C5D5N) δ158.2, 145.9, 137.1, 133.5, 130.8, 129.3, 128.5, 127.5, 125.8, 122.8, 120.4, 117.9, 117.7, 107.6, 71.8, 41.3, 38.5, 22.0, 19.6, 14.4. HRMS (LD+): m/z calculated for C20H18O2 [M]+ 290.1307, found 290.1297.
= +6.3(c 0.3, MeOH). IR (neat, cm−1): 3422, 2921, 2864, 1742, 1513, 1460, 1385. 1H NMR (400 MHz, Pyr) δ 12.00 (s, 1H), 8.64 (d, J=8.0 Hz, 1H), 8.57 (s, 1H), 7.94 (d, J=8.8 Hz, 1H), 7.84 (d, J=8.8 Hz, 1H), 7.64 (d, J=8.8 Hz, 1H), 7.52 (d, J=8.8 Hz, 1H), 6.98 (d, J=8.4 Hz, 1H), 6.51 (d, J=6.4 Hz, 1H), 4.86 (m, 1H), 3.87 (dd, J=16.8, 7.4 Hz, 1H), 3.61 (d, J=16.8 Hz, 1H), 1.46 (m, 1H), 1.36 (m, 1H), 1.30 (d, J=4.8 Hz, 3H), 1.16 (m, 1H). 13C NMR (100 MHz, C5D5N) δ158.2, 145.9, 137.1, 133.5, 130.8, 129.3, 128.5, 127.5, 125.8, 122.8, 120.4, 117.9, 117.7, 107.6, 71.8, 41.3, 38.5, 22.0, 19.6, 14.4. HRMS (LD+): m/z calculated for C20H18O2 [M]+ 290.1307, found 290.1297.
References
Machida, K. et al. Cinanthrenol A, an estrogenic steroid containing phenanthrene nucleus, from a marine sponge Cinachyrella sp. Org. Lett. 16, 1539–1541 (2014).
Ross, S. A. et al. Thaliporphinemethine: a new phenanthrene alkaloid from Illigera pentaphylla. J. Nat. Prod. 48, 835–836 (1985).
Hegde, R. V. et al. New potential antitumor compounds from the plant Aristolochia manshuriensis as inhibitors of the CDK2 enzyme. Bioorg. Med. Chem. Lett. 20, 1344–1346 (2010).
Behery, F. A. A. et al. Phenanthrenoids from Juncus acutus L., new natural lipopolysaccharide-inducible nitric oxide synthase inhibitors. Chem. Pharm. Bull. 55, 1264–1266 (2007).
Roddam, A. W., Allen, N. E., Appleby, P. & Key, T. J. Endogenous sex hormones and prostate cancer: a collaborative analysis of 18 prospective studies. J. Natl Cancer Inst. 100, 170–183 (2008).
DiGiovanni, J., Diamond, L., Harvey, R. G. & Slaga, T. J. Enhancement of the skin tumor-initiating activity of polycyclic aromatic hydrocarbons by methyl-substitution at non-benzo 'bay-region' positions. Carcinogenesis 4, 403–407 (1983).
Coombs, M. M., Bhatt, T. S. & Croft, C. J. Correlation between carcinogenicity and chemical structure in cyclopenta[α]phenanthrenes. Cancer Res. 33, 832–837 (1973).
Li, Z., Jin, Z. & Huang, R. Isolation, total synthesis and biological activity of phenanthroindolizidine and phenanthroquinolizidine alkaloids. Synthesis 16, 2365–2378 (2001).
Michael, J. P. Indolizidine and quinolizidine alkaloids. Nat. Prod. Rep. 18, 520–542 (2001).
Pschorr, R. Neue synthese des phenanthrens und seiner derivate. Chem. Ber. 29, 496–501 (1896).
Harvey, R. G. Polycyclic Aromatic Hydrocarbons, Wiley-VCH, New York, USA, (1997).
Zhang, Z., Sangaiah, R., Gold, A. & Ball, L. M. Synthesis of uniformly 13C-labeled polycyclic aromatic hydrocarbons. Org. Biomol. Chem. 9, 5431–5435 (2011).
Stoilov, I. et al. Synthesis of a triaromatic steroid biomarker, (20R, 24R-4, 17β-dimethyl-18, 19-dinorstigmasta-1, -3, -5, -7, -9, -11, -13-heptaene, from Stigmasterol. J. Org. Chem. 59, 926–928 (1994).
Coombs, M. M., Jaitly, S. B. & Crawley, F. E. H. Potentially carcinogenic cyclopenta[α]phenanthrenes. Part IV. Synthesis of 17-ketones by the Stobbe condensation. J. Chem. Soc. 1266–1271 (1970).
Coombs, M. M., Hall, M., Siddle, V. A. & Vose, C. W. Potentially carcinogenic cyclopenta[α]phenanthrenes. Part X. Oxygenated derivatives of the carcinogen 15, 16-dihydro-11-methylcyclopenta[α]phenanthren-17-one of metabolic interest. J. Chem. Soc. Perkin Trans. 1, 265–270 (1975).
Lee, H. & Harvey, R. G. New synthetic approaches to cyclopenta[α]phenanthrenes and their carcinogenic derivatives. J. Org. Chem. 53, 4253–4256 (1988).
Woski, S. A. & Koreeda, M. Synthesis of the putative active metabolites of the cyclopenta[α]phenanthrenes. Synthesis of the trans-3, 4-dihydro-3, 4-diol and syn-3, 4-diol 1, 2-epoxide derivatives of the mutagen 15, 16-dihydrocyclopenta[α]phenanthren-17-one. J. Org. Chem. 57, 5736–5741 (1992).
Furukawa, J., Kawabata, N. & Nishimura, J. Synthesis of cyclopropanes by the reaction of olefins with dialkylzinc and methylene iodide. Tetrahedron 24, 53–58 (1968).
Mallory, F. B., Wood, C. S. & Gordon, J. T. Photochemistry of stilbenes. III. Some aspects of the mechanism of photocyclization to phenanthrenes. J. Am. Chem. Soc. 86, 3094–3102 (1964).
Wang, K. -L., Lü, M. -Y., Wang, Q. -M. & Huang, R. -Q. Iron (III) chloride-based mild synthesis of phenanthrene and its application to total synthesis of phenanthroindolizidine alkaloids. Tetrahedron 64, 7504–7510 (2008).
Li, J., Yang, P., Yao, M., Deng, J. & Li, A. Total synthesis of rubriflordilactone A. J. Am. Chem. Soc. 136, 16477–16480 (2014).
Kaliakoudas, D., Eugster, C. H. & Ruedi, P. Synthese von Plectranthonen, diterpenoiden Phenanthren-1, 4-chinonen. Helv. Chim. Acta 73, 48–62 (1990).
Seçinti, H., Burmaoğlu, S., Altundaş, R. & Seçen, H. Total syntheses of multicaulins via oxidative photocyclization of stilbenes. J. Nat. Prod. 77, 2134–2137 (2014).
Sudhakar, A., Katz, T. J. & Yang, B. -W. Synthesis of a helical metallocene oligomer. J. Am. Chem. Soc. 108, 2790–2791 (1986).
Sudhakar, A. & Katz, J. J. Asymmetric synthesis of helical metallocenes. J. Am. Chem. Soc. 108, 179–181 (1986).
Hernandez-Perez, A. C., Vlassova, A. & Collins, S. K. Toward a visible light mediated photocyclization: Cu-based sensitizers for the synthesis of [5]Helicene. Org. Lett. 14, 2988–2991 (2012).
Li, H. et al. Straightforward synthesis of phenanthrenes from styrenes and arenes. Chem. Commun. 48, 7028–7030 (2012).
Hu, J. -Y. et al. Synthesis, structural, and helicenes. Eur. J. Org. Chem. 5829–5837 (2013).
Jørgensen, K. B. Photochemical oxidative cyclisation of stilbenes and stilbenoids-the Mallory-reaction. Molecules 15, 4334–4358 (2010).
Gore, P. H. & Kamonah, F. S. Photochemical synthesis of some monosubstituted chrysenes. Synthesis 773–775 (1978).
Rubottom, G. M., Vazquez, M. A. & Pelegrina, D. R. Peracid oxidation of trimethylsilyl enol ethers: a facile α-hydroxylation procedure. Tetrahedron Lett. 15, 4319–4322 (1974).
Davis, F. A., Sheppard, A. C., Chen, B. C. & Haque, M. S. Chemistry of oxaziridines. 14. Asymmetric oxidation of ketone enolates using enantiomerically pure (camphorylsulfonyl) oxaziridine. J. Am. Chem. Soc. 112, 6679–6690 (1990).
Ghanem, A. & Aboul-Enein, H. Y. Lipase-mediated chiral resolution of racemates in organic solvents. Tetrahedron Asymmetry 15, 3331–3351 (2004).
Lebel, H., Marcoux, J. -F., Molinaro, C. & Charette, A. B. Stereoselective cyclopropanation reactions. Chem. Rev. 103, 977–1050 (2003).
Acknowledgements
This research was financially supported by HKUST (R9309), Research Grant Council of Hong Kong (ECS 605912, GRF 605113, and GRF 16305314), and National Natural Science Foundation of China (NSFC 21472160).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Zhu, L., Tong, R. Total synthesis of ent-(+)-cinanthrenol A. J Antibiot 69, 280–286 (2016). https://doi.org/10.1038/ja.2015.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2015.114
