
Abstract
The ever-increasing bacterial resistance to clinical antibiotics is making many drugs ineffective and creating significant treatment gaps. This can be only circumvented by the discovery of antibiotics with new mechanisms of action. We report here the identification of a new tetramic acid, ascosetin, from an Ascomycete using the Staphylococcus aureus fitness test screening method. The structure was elucidated by spectroscopic methods including 2D NMR and HRMS. Relative stereochemistry was determined by ROESY and absolute configuration was deduced by comparative CD spectroscopy. Ascosetin inhibited bacterial growth with 2–16 μg ml−1 MIC values against Gram-positive strains including methicillin-resistant S. aureus. It also inhibited the growth of Haemophilus influenzae with a MIC value of 8 μg ml−1. It inhibited DNA, RNA, protein and lipid synthesis with similar IC50 values, suggesting a lack of specificity; however, it produced neither bacterial membrane nor red blood cell lysis. It showed selectivity for bacterial growth inhibition compared with fungal but not mammalian cells. The isolation, structure and biological activity of ascosetin have been detailed here.
Similar content being viewed by others
Introduction
Bacteria continue to acquire resistance against clinically used antibiotics making them less effective and posing a serious threat to human lives.1 Infections caused by methicillin-resistant Staphylococcus aureus (MRSA) alone are responsible for ∼18 000 deaths per year in the US.1 Although methicillin-resistant Staphylococcus aureus is well known and the subject of much attention, infections caused by a number of drug-resistant Gram-negative bacteria such as Pseudomonas aeruginosa and Acinetobacter baumannii are the cause for even greater concern. Most treatment options available today are the result of incremental improvements in the lead classes of antibiotics discovered during the Golden Age of antibiotic discovery. Unfortunately, further improvements to these classes of chemical structures are proving to be extremely challenging, leading to diminishing returns on investments. This may be surmounted by the discovery of new lead classes of antibacterial agents that can be developed for effective treatment of drug-resistant bacteria. It is well known that most of the antibacterial leads are of natural product origin.
Nature remains the most prolific source of antibacterial leads. There are many known natural antibacterials posing a challenge for dereplication and early differentiation of known from unknown compounds. To accomplish the goal of effective dereplication and for increasing the odds of finding novel compounds, we first implemented target-based whole-cell-sensitized S. aureus assays and then a genome-wide whole-cell antisense-based S. aureus fitness test.2 Screening with a fabF antisense strain led to the discovery of platensimycin3, 4 and platencin,5, 6 and an rpsD strain led to identification of several other target-based inhibitors.7, 8, 9, 10, 11 Recently, screening with S. aureus fitness test led to the discovery of kibdelomycins as gyrase B inhibitors.12, 13
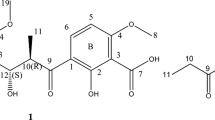
Continued screening using this approach allowed for the identification of a fungal extract that showed a novel, although complex, S. aureus fitness test profile and that produced no matches for known compounds by HPLC–HRESIFTMS (high-resolution electrospray ionization Fourier transformation mass spectrometry) analysis. The fermentation of this unidentified Ascomycete was scaled up for isolation and identification of the active component. Bioassay-guided fractionation of the extract using the S. aureus growth assay led to the isolation of a new tetramic acid, ascosetin (1, Figure 1). The isolation, structure elucidation and details of antibacterial activity including in vivo testing of this compound are described here.

Chemical structure of ascosetin.
Results and discussions
The producing microfungus, an unidentified Ascomycete, was isolated from a soil sample collected from the edge of a pond in Talamanca de Jarama, Madrid, Spain. It was grown on a solid millet-based medium for 29 days and extracted with an equal volume of acetone. After filtration, the extract was concentrated to remove most of the acetone and the aqueous layer was partitioned between water and ethyl acetate. Gel permeation (Sephadex LH20, GE Healthcare Biosciences, Pittsburgh, PA, USA) followed by two successive steps of reversed-phase C8 HPLC on the ethyl acetate extract afforded 35 mg (35 mg l−1) of ascosetin (1) as an amorphous powder.
Mass spectral analysis of 1 provided a molecular ion at m/z 444 (M+H). HRESIFTMS analysis produced a molecular formula of C26H37NO5. The UV spectrum of 1 showed absorption maxima at 220 and 290 nm. The IR spectrum showed absorption bands for hydroxyl groups (3223 cm−1) and conjugated ketones (1684 cm−1). The 13C NMR spectrum of 1 in CD3OD and DMSO-d6 showed the presence of 26 resonances, many of which were quite broadened (Table 1). These resonances were assigned to six methyls, four methylenes, two olefinic methines and six methines by a DEPT spectrum. The remaining resonances were quaternary in nature and were assignable to three olefinic, four keto and an sp3 aliphatic carbon. These assignments satisfied the molecular formula. The 13C NMR spectrum showed the presence of four resonances at δC 201.6, 180.3, 103.0 and 195.5 characteristic of the presence of a tetramic acid moiety.14 The 1H NMR spectrum of 1 showed three methyl doublets. A pair of these doublets was assigned to an isopropyl group by the COSY, TOCSY, 1H, 13C-HSQC and 1H, 13C HMBC spectral analyses. The remaining three methyl groups resonated as singlets, two of which could be assigned to olefinic methyls based on their 1H NMR shifts. The COSY and TOCSY analyses showed two large spin systems consisting of C5–C11–C18 and C5′–C9′. These spin systems were connected by HMBC correlations. The methyl group (H3-17) gave HMBC correlations to C-3 (δC 55.6), C-4 (δC 131.7) and C-5 (δC 129.3); H3-16 displayed correlations to C-3, C-13 (δC 161.3) and C-14 (δC 120.7); and H3-12 showed HMBC correlations to C-1 (δC 201.6), C-2 (δC 53.4) and C-11 (δC 42.4). These correlations, along with that of the correlation of H-14, to the carboxyl carbon at C-16 (δC 170.8) established the connectivity of carbons C1–C18. The HMBC correlations of H-5′ (δH 3.53) to C-2′ (δC 180.3) (both in CD3OD and DMSO-d6), together with those of the NH (δC 7.82) to C-3′ (δC 100.7), C-4′ (δC 195.5) and C-5′ (δC 59.1) in DMSO-d6, connected the tetramic acid moiety to the decalin moiety. The 1H, 15N-HSQC analysis showed a 1H and 15N cross-peak and confirmed the presence and assignment of NH (δH 7.82, s) to N (δN 112). Nitrogen showed 1H, 13N-HMBC correlation with H2–6′, thus confirming the presence of a tetramic acid moiety with an NH group.
The relative configuration of 1 was established by ROESY correlations in DMSO-d6 (Figure 2). Correlations between H-6 (δH 1.65), H-8 (δH 1.32) and H-10 (δH 0.55) suggest that these protons are present on the same face of the decalin moiety. Those of H-6 and H-8 to the methyl group H3-12 (δH 1.18) indicated that the methyl group is also on the same face. Correlations of H-11 (δH 1.58), H-7 (δH 0.70) and H-9 (δH 0.78) established them on the opposite face of the molecule. The ROESY analysis further established a chair conformation for the saturated ring and a flattened half-chair conformation for the monounsaturated ring of the trans-fused decalin with an equatorial methyl group at C-8 and an equatorial tetramic acid substituent at C-2 (Figure 2). The absolute configuration of ascosetin was assigned by comparison of the CD spectrum of 1 with the CD spectra of equisetin and the enantiomeric homolog phomasetin. The CD spectrum of 1 exhibited a negative cotton effect (Δɛ290=−6.97), which was similar in sign to the CD spectrum of (−)-equisetin (Δɛ290=−8.9) and opposite to that for (+)-phomasetin (Δɛ290=+5.2), confirming the absolute configuration of ascosetin as being identical to the configuration of (−)-equisetin.14 On the basis of these studies, the structure and absolute configuration of 1 was proposed for (−)-ascosetin. The configuration at C-5′ remains unassigned and has been shown to have no effect on the CD spectrum.14

ROESY correlations of 1 in DMSO-d6.
Ascosetin is likely derived from the condensation of polyketide and leucine nonribosomal peptide synthetase moieties. The methyl groups at C-8 and C-2 are known to have been derived from S-adenosyl methionine (SAM) in a related tetramic acid, trichosetin.15
Antibacterial activity
Ascosetin was tested for potency and spectrum of activity against a number of clinically relevant Gram-positive and Gram-negative bacteria. It showed growth inhibition of various strains of S. aureus with minimum inhibitory concentration (MIC) values of 2–8 μg ml−1 (Table 2). It inhibited the growth of Streptococcus pneumoniae with a MIC of 2 μg ml−1 in a iso-sensitest medium and showed strong media dependence. The activity of ascosetin against S. pneumoniae was significantly reduced when tested in the standard Mueller-Hinton medium containing 2.5% lysed horse blood (MIC 32 μg ml−1). It showed reasonable activity against Bacillus subtilis (MIC 8 μg ml−1) but was less active against Enterococcus faecalis (MIC 16 μg ml−1). Ascosetin was generally not active against Gram-negative bacteria (for example, Escherichia coli, whether wild-type or sensitized strain (envA/tolC) in which both outer membrane and efflux pumps are disrupted). However, it did show inhibition of Haemophilus influenzae (MIC 8 μg ml−1). The S. aureus inhibition was negatively affected by addition of 50% human serum, which produced a significant MIC shift (MIC>32 μg ml−1). Ascosetin did not inhibit the growth of Candida albicans.
To determine its mode of action, ascosetin was tested for its ability to inhibit the macromolecular synthesis of DNA, RNA, phospholipids, peptidoglycans and proteins in S. aureus EP167.3 In this assay, ascosetin exhibited no selectivity and inhibited all of the macromolecular synthetic pathways with almost equal potency (Figure 3) with IC50 values of 0.65 μg ml−1 (RNA), ∼1 μg ml−1 (DNA, proteins, phospholipids) and 1.50 μg ml−1 (peptidoglycans). Lack of selectivity suggests that ascosetin inhibits bacterial growth by a nonspecific manner but still maintains selectivity for inhibition of bacteria over fungi as evidenced by lack of inhibition of growth of C. albicans (MIC >64 μg ml−1). To further evaluate whether the activity exerted by ascosetin involved membrane lytic activity, it was tested in S. aureus bacterial (ATP release) and red blood cell lysis assays. Ascosetin did not have any effect in these two assays at 200 μM (highest concentration tested). In mammalian cytotoxicity assays, ascosetin showed IC50 values of 6, 5 and 10 μg ml−1, respectively, against the HeLa, CHO and HEK cell lines.

Macromolecular synthesis inhibition of S. aureus by ascosetin. A full color version of this figure is available at The Journal of Antibiotics journal online.
Ascosetin was tested for activity in a murine S. aureus target organ assay burden model of infection.3 It was not efficacious at 50 mg kg−1 by twice daily (100 mg kg−1 per day) subcutaneous dosing and produced visible signs of toxicity. It showed essentially flat exposure with area under the curve of 8/5/6 μM•h at 50/25/12.5 mg kg−1, respectively. Strong serum binding and low exposure likely affected the efficacy.
Tetramic acids with substituted decalin are widely distributed fungal metabolites with diverse biological effects, including antibacterial activity. Equisetin is perhaps the best known of the family; it was originally reported from Fusarium equiseti and subsequently isolated by us from F. heterosporum as an inhibitor of HIV-1 integrase.14 Interestingly, both enantiomers of this class of compounds are known to occur in nature. Phomasetin, the first enantiomeric homolog of equisetin, was reported from a Phoma species possessing similar potent HIV-1 integrase inhibitory activity.14 A number of tetramic acids have been reported to show antibacterial activities. These include trichosetin,16 equisetin and altersetin,17 CJ-21 058,18 CJ-17 57219 and cissetin.20 CJ-21 058 is also known to inhibit SecA (IC50 15 μg ml−1), an effector of ATP-dependent translocation of precursor proteins across bacterial cell membranes.18 Altersetin has been reported to exhibit weak in vivo activity in a murine sepsis model at 25 mg kg−1 by i.p. administration.17 Literature MIC values of tetramic acids against S. aureus are in the same range as those of ascosetin.
Experimental Section
General experimental procedures
Optical rotation was recorded on a Perkin-Elmer 241 polarimeter (Perkin-Elmer, Waltham, MA, USA), UV spectrum on a Perkin-Elmer Lambda 35 spectrometer and IR spectrum was recorded on a Perkin-Elmer Spectrum One FT-IR spectrophotometer. All NMR spectra were recorded with a Varian Unity 500 (Palo Alto, CA, USA) (1H, 500 MHz; 13C, 125 MHz) spectrometer in CD3OD or DMSO-d6 using a 3 mm NMR probe. Chemical shifts are reported in δ (p.p.m.) using residual solvent signals (DMSO-d6: δH 2.53 and δC 39.51 p.p.m.; CD3OD: δH 3.30 and δC 49.00) as internal standards. 1H, 13C, COSY, DQF-COSY, DEPT, HMQC, HMBC, TOCSY and NOESY spectra were recorded using standard Varian pulse sequences. Low-resolution mass spectrometry data were acquired on an Agilent 1100 MSD with ES ionization (Agilent, Santa Clara, CA, USA). HRESIFTMS was obtained on a Thermo Finnigan (Waltham, MA, USA) linear trap quadrupole (LTQ)-FTMS spectrometer. An Agilent HP 1100 instrument was used for analytical HPLC.
Producing organism and fermentation
The producing microfungus (E-000504855) was isolated from a soil sample collected at the edge of a pond in Talamanca de Jarama, Madrid, Spain. This is an Ascomycete, but the species remains unidentified. Fermentation was accomplished by inoculating several agar plugs with mycelia into a seed broth flask (50 ml medium in 250 ml unbaffled flask). The formulation for the seed media was as follows (gl−1), unless specified: corn steep liquor, 5.0; tomato paste, 40.0; oat flour, 10.0; glucose, 10.0; trace element mix, 10.0 ml l−1; agar, 4.0; pH adjusted to 6.8 with NaOH before sterilization. The formulation for the trace element mix consisted of (gl−1): FeSO4.7H20, 1.0; MnSO4 H20, 1.0; CuCl2·2H2O, 0.025; CaCl2, 0.1; H3BO3, 0.056; (NH4)6Mo6O24 4H2O, 0.019; ZnSO4 7H20, 0.2, prepared in 0.6 N HCl. The flasks were incubated at 22 °C with 80% relative humidity and shaken on a rotary shaker at 220 r.p.m.
After a seed stage of 4–7 days, a 1-ml aliquot was used to inoculate each flask of the production medium (50 ml medium in a 250-ml unbaffled flask). The formulation of the production media consisted of: millet, 15 g per flask; base liquid, 30.0 ml per flask. The formulation of the base liquid was as follows (gl−1): yeast extract, 33.3; sucrose, 33.3; alfalfa, 33.3; FeSO4·7H2O, 0.67; corn oil, 6.67; Na2C4H4O6·2H2O, 6.67. No pH adjustment was performed. The flasks were incubated at 22 °C with 80% relative humidity for 29 days before extraction.
Isolation of ascosetin (1)
One liter (20 flasks) of the solid state fermentation was extracted with 1 l of acetone and concentrated under reduced pressure to remove most of the acetone. The aqueous extract (250 ml) was extracted with two portions of 250 ml ethyl acetate. The combined ethyl acetate extracts were concentrated under reduced pressure to provide 1.7 g solid. This material was dissolved in 40 ml MeOH and 50% of the MeOH solution was charged to a 500 ml Sephadex LH20 column packed in and eluted with MeOH. The column was eluted at a flow rate of 5 ml min−1 and 50 ml fractions were collected. Fractions 29–36 showed antibacterial activity. They were pooled and concentrated to provide 120 mg of partially purified material. This material was dissolved in 500 μl MeOH and chromatographed in two equal portions on a reverse-phase Zorbax (Agilent) XDB C8 HPLC (150 × 21.2 mm) eluting with a 32-min gradient of 10–90% aqueous acetonitrile containing 0.1% trifluoroacetic acid at 10 ml min−1. Fractions 34–35 were pooled and lyophilized to provide 40 mg of almost pure ascosetin (1), which was re-purified on the same HPLC column and elution conditions, except this time without trifluoroacetic acid. Fractions 38–40 were pooled and lyophilized to afford 35 mg of ascosetin (1) (titer 35 mg l−1), as an amorphous powder. [α]23D −132.0 (c 0.5, MeOH); UV (MeOH) λmax 220 (log ɛ 3.73), 290 (3.57) nm; CD (MeOH) Δɛ290 −6.97; IR (ZnSe) νmax 3223, 2952, 2926, 2868, 1684, 1629, 1559, 1454, 1378, 1276, 1228, 1149, 1064 cm−1; electrospray ionization mass spectrometry m/z 444 [M+H]+; HRESIFTMS m/z 444.2745 (calcd for C26H37NO5+H, 444.2750), 1H and 13C NMR data, see Table 1.
MIC
The MIC against each of the strains was determined by the Clinical Laboratory Standards Institute guidelines by the twofold serial broth dilution method as previously described.21 Cultures were incubated at 37 °C for 20 h before activity was read. MIC is defined as the lowest concentration of antibiotic inhibiting visible growth.
Macromolecular synthesis inhibition
Macromolecular synthesis and labeling were performed in S. aureus EP167 using [14C-(U)]-glycine for peptidoglycan, [2-3H]-glycerol for phospholipid, [2-14C]-thymidine for DNA, [5, 6-3H]-uracil for RNA and L-[4, 5-3H]-leucine for protein following a procedure detailed earlier.22
In vivo efficacy
The in vivo efficacy studies were performed in a standard S. aureus target organ assay method as described earlier.3, 22 The compound was dissolved in DMSO and further diluted in sterile distilled water (10% DMSO final) and dosed subcutaneously at 50, 25 and 12.5 mg kg−1 twice daily.
Bacterial lysis assay
S. aureus (Smith) was grown in trypticase soy broth, subcultured into cation-adjusted Mueller-Hinton broth and grown to mid-log phase. The culture was diluted to a final OD600 of 0.1, dispensed into a 384-well microtiter plate and the test compounds diluted in DMSO were added. After mixing, samples were transferred to a 0.45-μm pore size filter plate (Multiscreen; EMD Millipore, Billerica, MA, USA) and incubated at 37 °C for 15 min. Following centrifugation at 3500 r.p.m. for 3 min, the filtrate was collected and ATP was quantitated using a coupled luciferase-based assay as described by the manufacturer (BacTiter-Glo Microbial Cell Viability Assay; Promega, Madison, WI, USA). Complete lysis was determined by the addition of 1% SDS. Data were analyzed using a 4-parameter curve-fitting algorithm.
Sheep red blood cell lysis assay
Sheep whole blood in Alsever’s solution (Becton Dickinson Diagnostic Systems, Franklin Lakes, NJ, USA) was washed in Tris-buffered saline, pH 7.4, diluted 1:40 and dispensed into a 384-well microtiter plate. Test compounds diluted in DMSO were added and the plate was incubated at 37 °C for 30 min. Following centrifugation at 3500 r.p.m. for 3 min, the cell-free supernatant was collected and hemoglobin was quantitated by measuring absorbance at 410 nm. Complete lysis was determined by the addition of 10% Triton-X (Sigma-Aldrich; St Louis, MO, USA). Data were analyzed using a 4-parameter curve-fitting algorithm.
HeLa cell cytotoxicity assay
The effects of test compounds on eukaryotic cell DNA synthesis and growth were assessed using the modified thymidine analog 5-ethynyl-2′-deoxyuridine (EdU) fluorescently labeled with Alexa Fluor 488 dye through click chemistry as described by the manufacturer (Click-iT EdU Alexa Fluor 488 HCS Assay; Invitrogen, Grand Island, NY, USA). Human cervical adenocarcinoma cells (HeLa; ATCC, Manassas, VA, USA) were used and DNA synthesis as well as total cell counts were assessed following incubation with test compounds and 5 μM EdU at 37 °C for 24 h. Cells were fixed and EdU was click labeled with Alexa Fluor 488 azide. Microplates were analyzed on an Acumen eX3 laser scanning plate cytometer (TTP Labtech; Melbourne, UK). Data were analyzed using a 4-parameter curve-fitting algorithm.
References
Klevens, R. M. et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. J. Am. Med. Assoc. 298, 1763–1771 (2007).
Donald, R. G. et al. A Staphylococcus aureus fitness test platform for mechanism-based profiling of antibacterial compounds. Chem. Biol. 16, 826–836 (2009).
Wang, J. et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 441, 358–361 (2006).
Singh, S. B. et al. Isolation, structure, and absolute stereochemistry of platensimycin, a broad spectrum antibiotic discovered using an antisense differential sensitivity strategy. J. Am. Chem. Soc. 128, 11916–11920 and 15547 (2006).
Wang, J. et al. Platencin is a dual fabf and fabh inhibitor with potent in vivo antibiotic properties. Proc. Natl Acad. Sci. USA 104, 7612–7616 (2007).
Jayasuriya, H. et al. Isolation and structure of platencin: a novel FabH and FabF dual inhibitor with potent broad spectrum antibiotic activity produced by streptomyces platensis MA7339. Angew. Chem. Int. Ed. Engl. 46, 4684–4688 (2007).
Singh, S. B. et al. Discovery of lucensimycins A and B from Streptomyces lucensis MA7349 using an antisense strategy. Org. Lett. 8, 5449–5452 (2006).
Ondeyka, J. G. et al. Coniothyrione, a chlorocyclopentandienylbenzopyrone as a bacterial protein synthesis inhibitor discovered by antisense technology. J. Nat. Prod. 70, 668–670 (2007).
Zhang, C. et al. Isolation, structure and antibacterial activity of pleosporone from a pleosporalean ascomycete discovered by using antisense strategy. Bioorg. Med. Chem. 17, 2162–2166 (2009).
Zhang, C. et al. Isolation, structure and antibacterial activity of phaeosphenone from a Phaeosphaeria sp. discovered by antisense strategy. J. Nat. Prod. 71, 1304–1307 (2008).
Singh, S. B., Zink, D. L., Herath, K. B., Salazar, O. & Genilloud, O. Discovery and antibacterial activity of lucensimycin C from Streptomyces lucensis. Tetrahedron Lett. 49, 2616–2619 (2008).
Phillips, J. W. et al. Discovery of kibdelomycin, a potent new class of bacterial type II topoisomerase inhibitor by chemical-genetic profiling in Staphylococcus aureus. Chem. Biol. 18, 955–965 (2011).
Singh, S. B. et al. Kibdelomycin A, a congener of kibdelomycin, derivatives and their antibacterial activities. Bioorg. Med. Chem. Lett. 22, 7127–7130 (2012).
Singh, S. B. et al. Equisetin and a novel opposite stereochemical homolog phomasetin, two fungal metabolites as inhibitors of HIV-1 integrase. Tetrahedron Lett. 39, 2243–2246 (1998).
Marfori, E. C., Bamba, T., Kajiyama, S. i., Fukusaki, E. -i. & Kobayashi, A. Biosynthetic studies of the tetramic acid antibiotic trichosetin. Tetrahedron 58, 6655–6658 (2002).
Marfori, E. C., Kajiyama, S., Fukusaki, E. & Kobayashi, A. Trichosetin, a novel tetramic acid antibiotic produced in dual culture of Trichoderma harzianum and Catharanthus roseus Callus. Z. Naturforsch. C 57, 465–470 (2002).
Hellwig, V. et al. Altersetin, a new antibiotic from cultures of endophytic Alternaria spp. Taxonomy, fermentation, isolation, structure elucidation and biological activities. J. Antibiot. (Tokyo) 55, 881–892 (2002).
Sugie, Y. et al. CJ-21,058, a new SecA inhibitor isolated from a fungus. J. Antibiot. (Tokyo) 55, 25–29 (2002).
Sugie, Y. et al. A novel antibiotic CJ-17,572 from a fungus, Pezicula sp. J. Antibiot. (Tokyo) 55, 19–24 (2002).
Boros, C., Dix, A., Katz, B., Vasina, Y. & Pearce, C. Isolation and identification of cissetin—a setin-like antibiotic with a novel cis-octalin ring fusion. J. Antibiot. (Tokyo) 56, 862–865 (2003).
Onishi, H. R. et al. Antibacterial agents that inhibit lipid A biosynthesis. Science 274, 980–982 (1996).
Zhang, C. et al. Isolation, structure, and antibacterial activity of philipimycin, a thiazolyl peptide discovered from Actinoplanes philippinensis MA7347. J. Am. Chem. Soc. 130, 12102–12110 (2008).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article
Cite this article
Ondeyka, J., Smith, S., Zink, D. et al. Isolation, structure elucidation and antibacterial activity of a new tetramic acid, ascosetin. J Antibiot 67, 527–531 (2014). https://doi.org/10.1038/ja.2014.33
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2014.33
Keywords
This article is cited by
-
Exploring a general multi-pronged activation strategy for natural product discovery in Actinomycetes
Communications Biology (2024)
-
Recent advances in the chemo-biological characterization of decalin natural products and unraveling of the workings of Diels–Alderases
Fungal Biology and Biotechnology (2022)
