
Abstract
An alternative and improved total synthesis of incednam, the aglycon of the 24-membered macrolactam glycoside antibiotic incednine, was accomplished. The synthesis was realized via construction of the 24-membered macrocycle using intramolecular ring-closing olefin metathesis reaction as a key step.
Similar content being viewed by others

Introduction
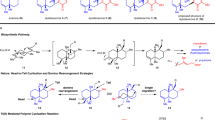
Incednam (1) is the aglycon of the 24-membered macrolactam glycoside antibiotic incednine (2), which was isolated by Imoto et al.1 in 2008. Compound 2 exhibits significant inhibitory activity against the anti-apoptotic oncoproteins Bcl-2 and Bcl-xL, with a mode of action distinctly different from those of other agents that inhibit the binding capacity of Bcl-xL to the pro-apoptotic protein Bax. Furthermore, these proteins are overexpressed in many cancer cells, resulting in the expansion of a transformed population and promotion of the multidrug-resistant stage.2, 3, 4 Therefore, 2 is expected to be a lead compound in the development of novel antitumor drugs. In addition, 2 is likely to be a useful tool for the further study of Bcl-2 and Bcl-xL functions. The identification of its target protein could provide insight into the anti-apoptotic mechanism of the Bcl-2 family proteins. From a chemical structural perspective, 1 and 2 contain unique features: an α-methoxy-α,β-unsaturated amide moiety and two independent conjugated polyene systems embedded in the 24-membered macrolactam ring. Due to the nature of the highly conjugated polyene subunits, 1 and 2 are light- and acid-sensitive. Although 1 was also isolated from Streptomyces sp.,1 its semi-synthesis from 2 has not been realized, in part due to its inherent chemical instabilities. However, their important biological activities and novel molecular architecture make 1 and 2 prime targets for chemical synthesis. The first total synthesis of 1 involved preparation of the C1–C13 subunit 3 and the C14–C23 subunit 4, and construction of the novel 24-membered macrocycle through Stille coupling between 3 and 4, followed by macrolactamization as shown in Figure 1.5 The present report describes an alternative and improved synthesis of 1 via construction of the 24-membered macrocycle by intramolecular ring-closing olefin metathesis reaction as a key step.

Retrosynthetic analysis of incednam (1).
Results and Discussion
The initial total synthesis of 1 was accomplished by preparation of the C14–C23 subunit 4, which could not be stored and was used immediately for the next step due to the instability.5 To circumvent this issue for the practical synthesis of 1, the retrosynthesis of 1 was redesigned via precursor 5 or 6 for intramolecular ring-closing olefin metathesis reaction6, 7, 8 to construct the 24-membered macrocycle concomitant with the labile C14-C21 tetraene unit at a later stage in the synthesis. The new retrosynthetic analysis of 1 is depicted in Figure 1. The convergent strategy applied to the construction of the 24-membered macrocycle is based on coupling of three domains: the C1–C13 subunit 3 containing the vinyl iodide moiety,5 the C14–C18 subunit 7 containing the vinyl stannane moiety, and the C19–C23 subunit 8 containing the amino group. This union was produced by application of intermolecular Stille coupling and amidation, followed by intramolecular ring-closing olefin metathesis reaction.
The synthesis of the triene subunit 7, corresponding to the C14–C18 in 1, is summarized in Scheme 1. The known aldehyde 10 was prepared from ethyl 2-butynoate (9) in 5 steps by a procedure previously reported.9, 10 Wittig reaction of 10 with Ph3P=CH2 in CH2Cl2 provided the triene 7 in 88% yield. Next, the synthesis of the C19–C23 subunit 8 was accomplished starting from the alcohol 12, which was prepared as in the initial total synthesis of 1,5 as shown in Scheme 2. The secondary alcohol 12 was converted into the mesylate 13 utilizing methanesulfonyl chloride (MsCl) in pyridine (Py), which was subsequently transformed into the azide 14 using NaN3 in DMF at 110 °C with stereochemical inversion in 88% overall yield. Deprotection of the tert-butyldiphenylsilyl (TBDPS) ether of 14 with tetrabutylammonium fluoride (TBAF) in THF, and subsequent oxidation of the resulting allyl alcohol 15 using MnO2 in CH2Cl2 provided the aldehyde 16 in 89% overall yield. Wittig reaction of 16 using Ph3P=CH2 gave the diene 17 in 72% yield. Finally, reduction of the azide group of 17 using PPh3 under Staudinger’s conditions11, 12 furnished the amine 8 in 98% yield.
With the key fragments 7 and 8 in hand, attention turned to the total synthesis of 1 using 3. Completion of the synthesis of 1 is summarized in Scheme 3. Amidation of 3 and 8 using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 4-dimethylaminopyridine (DMAP) in CH2Cl2 proceeded smoothly to give the amide 18 in 94% yield. Subsequently, Stille coupling of 18 and 7 using Pd2(dba)3 in the presence of LiCl and CuCl in DMF/THF13 gave the best result, providing the desired coupling product 19 in 71% yield. Removal of the triethylsilyl (TES) groups of 19 using TBAF and AcOH in THF furnished the diol 20 in 42% yield. Conditions for the intramolecular ring-closing olefin metathesis of 19 or 20 were rigorously explored using Grubbs first-generation,14 Grubbs second-generation,15 Hoveyda–Grubbs first-generation,16 Hoveyda–Grubbs second-generation,17 and Grela18, 19 catalysts. Experimentation revealed that the best conditions were those using 19 and Grela catalyst 21 in the presence of P-methoxyphenol (PMPOH) and MS 3A in CH2Cl2 to give incednam (1) in 17% overall yield after deprotection of the TES groups in the resulting cyclic product. 1H-NMR, 13C-NMR, HRMS (ESI-TOF), and optical rotation data obtained for a sample of the synthetic incednam matched those of an authentic sample1 and of a sample from the initial synthesis of 1.5
In conclusion, a novel convergent synthetic route was developed for incednam (1), which is the aglycon of the 24-membered macrolactam glycoside antibiotic incednine (2), using intramolecular ring-closing olefin metathesis reaction as a key step. Although the yield of the intramolecular ring-closing olefin metathesis reaction was not extremely high, the present synthetic route avoids the use of unstable fragments, such as 4, in the total synthesis of 1. Furthermore, this approach shows potential for intramolecular ring-closing olefin metathesis even in a complex structure possessing polyene units. Additional studies related to the total synthesis of incednine (2) from 1 are currently underway.20
With great respect, we dedicate this work to Professor Kuniaki Tatsuta as a memorial to his total synthesis of 101 antibiotics. This research was supported in part by the MEXT-supported Program for the Strategic Research Foundation at Private Universities, 2012–2016, Scientific Research on Innovative Areas ‘Chemical Biology of Natural Products’ and JSPS Fellow 22·5820 from MEXT.
Experimental procedure
Melting points were determined on a micro hot-stage (Yanako MP-S3) and were uncorrected. Optical rotations were measured on a JASCO P-2200 polarimeter. 1H-NMR spectra were recorded on a JEOL ECA-500 (500 MHz) spectrometer. 13C-NMR spectra were taken on a JEOL ECA-500 (125 MHz) spectrometer in CDCl3 at room temperature, unless otherwise noted. 1H-NMR data were reported as follows: chemical shift in parts par million (p.p.m.) downfield or upfield from tetramethylsilane (δ 0.00), CHCl3 (δ 7.26), integration, multiplicity (br=broad, s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet) and coupling constants (Hz). 13C chemical shifts were reported in p.p.m. downfield or upfield from CDCl3 (δ 77.36) or acetone-d6 (δ 30.60). ESI-TOF Mass spectra and APCI-TOF Mass spectra were measured on a Waters LCT premier XE. Silica-gel TLC and column chromatography were performed on Merck TLC 60F-254 (0.25 mm) and Kanto Chemical Co., Inc. (Tokyo, Japan) Silica-Gel 60 N (spherical, neutral), respectively. Air- and/or moisture-sensitive reactions were carried out under an atmosphere of argon using oven-dried glassware. In general, organic solvents were purified and dried using an appropriate procedure, and evaporation and concentration were carried out under reduced pressure below 30 °C, unless otherwise noted.

Synthesis of the C14–C18 subunit 7.

Synthesis of the C19–C23 subunit 8.

Total synthesis of incednam (1).
(1E,3E,5E)-3-Methylhex-1,3,5-trien-1-tributylstannane (7)
To a solution of 10 (56.6 mg, 146 μmol) in dry CH2Cl2 (1.12 ml) was added Ph3P=CH2 (202 mg, 731 μmol) under Ar atmosphere at room temperature. The reaction mixture was stirred for 15 h at the temperature, the mixture was quenched by addition of a saturated NH4Cl aq. (1 ml). The resulting mixture was extracted with CHCl3 (2 ml × 3). The combined organic layer was washed with brine (1 ml), dried over anhydrous Na2SO4, and concentrated in vacuo. Purification of the residue by column chromatography (hexane) on aluminum oxide activated, basic, Brockmann I gave 7 (49.0 mg, 128 μmol, 88% yield). Pale yellow syrup; Rf 0.72 (10/1 hexane/EtOAc); 1H-NMR (CDCl3, TMS) δ 6.71 (1H, ddd, J=10.0, 11.2, 16.9 Hz), 6.57 (1H, d, J=19.2 Hz), 6.30 (1H, d, J=19.2 Hz), 6.06 (1H, d, J=11.2 Hz), 5.26 (1H, d, J=16.9 Hz), 5.13 (1H, d, J=10.0 Hz), 1.87 (3H, s), 1.50 (6H, m), 1.31 (6H, m), 0.90 (15H, m); 13C-NMR (CDCl3) δ 150.7, 137.2, 133.6, 131.4, 128.0, 117.8, 29.3, 27.5, 13.9, 12.2, 9.7; HRMS (ESI-TOF) m/z 385.19 (385.1921 calcd for C19H37Sn, [M+Na]+).
(2E,5R)-1-tert-Butyldiphenylsilyloxy-5-methanesulfonyloxy-2-methylhex-2-ene (13)
To a solution of 12 (1.30 g, 3.53 mmol) in dry pyridine (19.5 ml) was added MsCl (410 μl, 5.30 mmol) under Ar atmosphere at 0 °C. After the mixture was stirred for 3 h at room temperature, the reaction was quenched by addition of water (20 ml). The resulting mixture was extracted with EtOAc (10 ml × 3). The combined organic layer was washed with brine (20 ml), dried over anhydrous Na2SO4, and concentrated in vacuo. Purification of the residue by silica-gel column chromatography (hexane/EtOAc=4/1) gave 13 (1.53 g, 3.42 mmol, 97% yield). Colorless syrup; Rf 0.60 (2/1 hexane/EtOAc); [α]26D +2.4° (c 0.28, CHCl3); 1H-NMR (CDCl3, TMS) δ 7.65 (4H, dd, J=1.5, 7.7 Hz), 7.39 (6H, m), 5.51 (1H, dt, J=1.7, 7.5 Hz), 4.79 (1H, ddq, J=6.3, 6.3, 6.6 Hz), 4.06 (2H, s), 2.94 (3H, s), 2.52 (1H, ddd, J=6.6, 7.5, 14.0 Hz), 2.38 (1H, ddd, J=6.3, 7.5, 14.0 Hz), 1.61 (3H, s), 1.41 (3H, d, J=6.3 Hz) 1.06 (9H, s); 13C-NMR (CDCl3) δ 138.2, 135.6, 133.7, 129.8, 127.8, 117.4, 80.0, 68.4, 38.6, 34.8, 26.9, 21.1, 19.4, 13.9; Anal. calcd for C24H34O4SSi: C, 64.53; H, 7.67; S, 7.18. Found: C, 64.37; H, 7.85; S, 7.46.
(2E,5S)-5-Azido-1-tert-butyldiphenylsilyloxy-2-methylhex-2-ene (14)
To a solution of 13 (1.53 g, 3.41 mmol) in dry DMF (20.0 ml) was added NaN3 (333 mg, 5.12 mmol) under Ar atmosphere at room temperature. After the mixture was stirred for 1.5 h at 110 °C, the reaction was quenched by addition of water (20 ml). The resulting mixture was extracted with hexane/EtOAc=1/1 (10 ml × 3). The combined organic layer was washed with brine (20 ml), dried over anhydrous Na2SO4, and concentrated in vacuo. Purification of the residue by silica-gel column chromatography (hexane/EtOAc=20/1) gave 14 (1.22 g, 3.10 mmol, 91% yield). Colorless syrup; Rf 0.61 (5/1 hexane/EtOAc); [α]26D +6.6° (c 0.28, CHCl3); 1H-NMR (CDCl3, TMS) δ 7.67 (4H, d, J=6.6 Hz), 7.39 (6H, m), 5.50 (1H, t, J=7.5 Hz), 4.07 (2H, s), 3.48 (1H, tq, J=6.6, 6.9 Hz), 2.31 (1H, ddd, J=6.9, 7.5, 14.1 Hz), 2.22 (1H, ddd, J=6.9, 7.5, 14.1 Hz), 1.61 (3H, s), 1.23 (3H, d, J=6.6 Hz), 1.07 (9H, s); 13C-NMR (CDCl3) δ 137.3, 135.6, 133.9, 129.7, 127.7, 119.1, 77.4, 77.1, 76.9, 68.7, 58.0, 34.3, 26.9, 19.4, 19.2, 13.8; Anal. Calcd for C23H31N3OSi: C, 70.18; H, 7.94. Found: C, 69.85; H, 8.00.
(2E,5S)-5-Azido-2-methylhex-2-en-1-ol (15)
To a solution of 14 (1.91 g, 4.85 mmol) in dry THF (38.0 ml) was added 1.0 M TBAF in THF (7.27 ml, 7.27 mmol) under Ar atmosphere at 0 °C. After the mixture was stirred for 3 h at room temperature, the reaction was quenched by addition of H2O (10 ml). The resulting mixture was extracted with EtOAc (20 ml × 3). The combined organic layer was washed with brine (10 ml), dried over anhydrous Na2SO4, and concentrated in vacuo. Purification of the residue by silica-gel column chromatography (hexane/EtOAc=2/1, 1% Et3N) gave 15 (726 mg, 4.68 mmol, 96% yield). Colorless syrup; Rf 0.18 (5/1 hexane/EtOAc); [α]25D +11.9° (c 0.49, CHCl3); 1H-NMR (CDCl3, TMS) δ 5.45 (1H, dt, J=1.6, 7.2 Hz), 4.03 (2H, s), 3.51 (1H, m), 2.27 (2H, m), 1.69 (3H, s), 1.26 (3H, d, J=6.6 Hz); 13C-NMR (CDCl3) δ 138.1, 120.7, 68.5, 34.5, 19.3, 14.0; HRMS (ESI-TOF) m/z 128.1077 (128.1075 calcd for C7H14NO, [MH-N2]+).
(2E,5S)-5-Azido-2-methylhex-2-en-1-al (16)
To a solution of 15 (726 mg, 4.68 mmol) in dry CH2Cl2 (46.8 ml) was added MnO2 (4.07 g, 46.8 mmol) under Ar atmosphere at the room temperature. After the mixture was stirred for 15 h at 40 °C, the mixture was filtered through a pad of Celite. The combined filtrates were concentrated in vacuo. Purification of the residue by silica-gel column chromatography (hexane/EtOAc=2/1) gave 16 (663 mg, 4.33 mmol, 93% yield). Colorless syrup; Rf 0.33 (5/1 hexane/EtOAc); [α]24D +24.3° (c 0.68, CHCl3); 1H-NMR (CDCl3, TMS) δ 9.45 (1H, s), 6.51 (1H, dt, J=1.4, 7.2 Hz), 3.71 (1H, m), 2.55 (2H, dd, J=6.6, 7.0 Hz), 1.78 (3H, s), 1.35 (3H, d, J=6.6 Hz); 13C-NMR (CDCl3) δ 194.9, 148.5, 141.5, 56.8, 35.5, 19.5, 9.6; HRMS (ESI-TOF) m/z 126.0917 (126.0919 calcd for C7H12NO, [MH-N2]+).
(2E,4E,6E)-2-Azido-5-methylhepta-4,6-diene (17)
To a solution of 16 (175 mg, 1.14 mmol) in dry Et2O (11.4 ml) was added Ph3P=CH2 (450 mg, 1.60 mmol) under Ar atmosphere at 0 °C. The reaction mixture was stirred for 1 h at the room temperature, the mixture was quenched by addition of a saturated NH4Cl aq. (5 ml). The resulting mixture was extracted with Et2O (5 ml × 3). The combined organic layer was washed with brine (5 ml), dried over anhydrous Na2SO4, and concentrated in vacuo. Purification of the residue by silica-gel column chromatography (hexane/Et2O=30/1, 1% Et3N) gave 17 (124 mg, 822 μmol, 72% yield). Pale yellow syrup; Rf 0.71 (10/1 hexane/EtOAc); [α]25D +12.1° (c 0.52, CHCl3); 1H-NMR (CDCl3, TMS) δ 6.38 (1H, dd, J=10.6, 17.5 Hz), 5.48 (1H, t, J=7.2 Hz), 5.14 (1H, d, J=17.5 Hz), 4.99 (1H, d, J=10.6 Hz), 3.52 (1H, m), 2.35 (2H, m), 1.77 (3H, s), 1.26 (3H, d, J=6.6 Hz); 13C-NMR (CDCl3) δ 141.2, 136.8, 127.5, 111.9, 57.9, 35.0, 19.3, 12.1; HRMS (ESI-TOF) m/z 124.1127 (124.1126 calcd for C8H14N, [MH−N2]+).
(2E,4E,6E)-2-Amino-5-methylhepta-4,6-diene (8)
To a solution of 17 (124 mg, 822 μmol) in THF/H2O (10/1, v/v, 11.7 ml) was added PPh3 (431 mg, 1.64 mmol) under Ar atmosphere at room temperature. After the mixture was stirred for 15 h at 40 °C, the mixture was quenched by addition of H2O (5 ml). The resulting mixture was extracted with Et2O (5 ml × 3). The combined organic layer was washed with brine (5 ml), dried over anhydrous Na2SO4, and concentrated in vacuo. Purification of the residue by silica-gel column chromatography (CHCl3/MeOH=10/1–5/1, 1% NH3 aq.) gave 8 (101 mg, 806 μmol, 98% yield). Pale yellow syrup; Rf 0.23 (5/1 CHCl3/MeOH); [α]24D +8.21° (c 0.84, CHCl3); 1H-NMR (CDCl3, TMS) δ 6.39 (1H, dd, J=10.6, 17.5 Hz), 5.51 (1H, t, J=7.5 Hz), 5.11 (1H, d, J=17.5 Hz), 4.95 (1H, d, J=10.6 Hz), 2.99 (1H, m), 2.19 (2H, dd, J=7.2, 7.5 Hz), 1.74 (3H, s), 1.42 (2H, s), 1.09 (3H, d, J=6.3 Hz); 13C-NMR (CDCl3) δ 141.5, 136.0, 129.9, 111.1, 47.4, 38.9, 23.7, 12.0; HRMS (ESI-TOF) m/z 126.1283 (126.1283 calcd for C8H16N, [M+H]+).
(2Z,4E,6E,8E,10R,11S,12Z)-1-((2E,4E,6E)-5-Methylhepta-4,6-diene)amide-10,11-bis(triethylsilyloxy)-13-iodo-2-methoxy-4,10,16-trimethyltrideca-2,4,6,8,12-pentaene (18)
To a solution of 3 (114 mg, 176 μmol) in CH2Cl2 (1.50 ml) were added DMAP (43.0 mg, 352 μmol) and EDC (67.4 mg, 352 μmol) under Ar atmosphere at 0 °C. After the mixture was stirred for 20 min at the temperature, a solution of 8 (88.1 mg, 704 μmol) in CH2Cl2 (2.00 ml) was added and stirring was continued for another 13 h at room temperature. The reaction was quenched by addition of H2O (2 ml). The resulting mixture was extracted with EtOAc (2 ml × 3). The combined organic layer was washed with brine (2 ml), dried over anhydrous Na2SO4, and concentrated in vacuo. Purification of the residue by silica-gel column chromatography (hexane/EtOAc=10/1 to 4/1, 1% Et3N) gave 18 (115 mg, 152 μmol, 94% yield). Pale yellow syrup; Rf 0.30 (5/1 hexane/EtOAc); [α]26D +23.7° (c 0.89, CHCl3); 1H-NMR (CDCl3, TMS) δ 6.67 (1H, s), 6.51 (1H, dd, J=11.5, 14.0 Hz), 6.40 (1H, d, J=11.5 Hz), 6.38 (1H, dd, J=10.9, 17.5 Hz), 6.33 ( 1H, dd, J=10.9, 14 Hz), 6.32 (1H, d, J=7.7 Hz), 6.24 (1H, dd, J=10.9, 15.2 Hz), 6.15 (1H, dd, J=7.7, 8.3 Hz), 5.86 (1H, d, J=15.2 Hz), 5.50 (1H, t, J=7.5 Hz), 5.12 (1H, d, J=17.5 Hz), 4.97 (1H, d, J=10.9 Hz), 4.14 (1H, m), 4.12 (1H, d, J=8.3 Hz), 3.57 (3H, s), 2.38 (2H, dd, J=6.9, 7.5 Hz), 2.09 (3H, s), 1.77 (3H, s), 1.35 (3H, s), 1.20 (3H, d, J=6.6 Hz), 0.93 (18H, m), 0.59 (12H, m); 13C-NMR (CDCl3) δ 163.7, 147.3, 141.3 × 2, 140.8, 136.7, 135.9, 135.4, 131.8, 129.6, 128.4, 128.1, 124.8, 111.6, 83.6, 81.3, 78.3, 61.1, 45.5, 35.2, 22.9, 20.6, 15.0, 12.1, 7.3, 7.0, 6.8, 5.2; HRMS (ESI-TOF) m/z 756.3340 (756.3340 calcd for C36H63NO4Si2I, [M+H]+).
(2Z,4E,6E,8E,10R,11S,12Z,14E,16E,18E)-1-((2E,4E,6E)-5-Methylhepta-4,6-diene)amide-10,11-bis(triethylsilyloxy)-13-iodo-2-methoxy-4,10,16-trimethylnonadeca-2,4,6,8,12,14,16-heptaene (19)
To a solution of 18 (25.8 mg, 34.1 μmol) and 7 (78.5 mg, 205 μmol) in dry THF/DMF (1/1, v/v, 683 μl) were added LiCl (11.6 mg, 273 μmol), CuCl (20.3 mg, 205 μmol) and Pd2(dba)3 (6.3 mg, 6.83 μmol) under Ar atmosphere at room temperature. After the mixture was stirred for 4 h, the reaction was quenched by addition of saturated NaHCO3 aq. (1 ml). The resulting mixture was extracted with hexane/EtOAc (1/1, v/v, 1 ml × 3). The combined organic layer was washed with brine (5 ml), dried over anhydrous Na2SO4, and concentrated in vacuo. Purification of the residue by silica-gel column chromatography (hexane/EtOAc=10/1 to 8/1, 1% Et3N) gave 19 (16.5 mg, 22.8 μmol, 71% yield). Pale yellow syrup: Rf 0.31 (5/1 hexane/EtOAc); [α]26D − 86.8° (c 0.98, CHCl3); 1H-NMR (CDCl3, TMS) δ 6.72 (1H, ddd, J=10.0, 11.2, 16.6 Hz), 6.56 (1H, dd, J=11.2, 15.2 Hz), 6.51 (1H, dd, J=11.5, 14.1 Hz), 6.41 (1H, d, J=11.5 Hz), 6.37 (1H, dd, J=11.2, 17.5 Hz), 6.32 (1H, dd, J=11.8, 14.1 Hz), 6.26 (1H, d, J=15.2 Hz), 6.25 (1H, dd, J=11.8, 14.9 Hz), 6.12 (1H, d, J=11.2 Hz), 6.11 (1H, dd, J=10.3, 11.2 Hz), 5.91 (1H, d, J=14.9 Hz), 5.50 (1H, t, J=7.2 Hz), 5.37 (1H, dd, J=9.2, 10.3 Hz), 5.26 (1H, d, J=16.6 Hz), 5.15 (1H, d, J=10.0 Hz), 5.12 (1H, d, J=17.5 Hz), 4.97 (1H, d, J=11.2 Hz), 4.32 (1H, d, J=9.2 Hz), 4.14 (1H, m), 3.57 (3H, s), 2.38 (2H, dd, J=6.9, 7.2 Hz), 2.09 (3H, s), 1.90 (3H, s), 1.77 (3H, s), 1.31 (3H, s), 1.20 (3H, d, J=6.6 Hz), 0.91 (18H, m), 0.59 (12H, m); 13C-NMR (CDCl3) δ 163.7, 147.3, 142.1, 141.3, 138.3, 136.7, 136.0, 135.9, 135.6, 133.3, 132.3, 131.7, 131.6, 130.8, 129.2, 128.2, 128.1 × 2, 125.0, 124.8, 117.8, 111.6, 61.1, 45.5, 35.2, 21.8, 20.6, 15.0, 12.8, 12.1, 7.4, 7.3, 7.0, 6.8, 5.1; HRMS (ESI-TOF) m/z 722.4979 (722.5000 calcd for C43H72NO4Si2, [M+H]+).
Incednam (1)
To a stirred solution of 19 (16.6 mg, 23.0 μmol) in deaerated CH2Cl2 (12.0 ml) was added molecular sieves 3A (16.6 mg), p-methoxyphenol (5.7 mg, 46.0 μmol) and Grela catalyst 21 (6.2 mg, 9.20 μmol) at room temperature. After stirring at the temperature for 6.5 h, the solution was passed through silica-gel column chromatography (CHCl3, 1% Et3N), and concentrated in vacuo. The crude product (3.9 mg) was used for the next reaction without further purification. To a solution of crude product (3.9 mg) in dry THF (562 μl) were added the mixture of 1.0 M TBAF in THF (33.7 μl, 33.7 μmol) and AcOH (1.6 μl, 28.1 μmol) under Ar atmosphere. The reaction mixture was stirred for 16 h at room temperature, and then the mixture of 1.0 M TBAF in THF (67.4 μl, 67.4 μmol) and AcOH (3.2 μl, 56.2 μmol) was added at the same temperature. After the mixture was stirred for 8 h at the temperature, the reaction was quenched by addition of saturated NaHCO3 aq. (2 ml). The resulting mixture was extracted with EtOAc (2 ml × 3). The combined organic layer was washed with brine (2 ml), dried over anhydrous Na2SO4, and concentrated in vacuo. Purification of the residue by silica-gel column chromatography (CHCl3/MeOH=40/1, 1% Et3N) gave 1 (1.8 mg, 3.9 μmol, 17% yield in two steps). Data for an analytical sample of the synthetic incednam (1) obtained by 1H-NMR, HRMS (ESI-TOF) and optical rotation matched those obtained for an authentic sample and a sample from the 1st generation synthesis.1, 5 Pale yellow powder; Rf 0.46 (10/1 CHCl3/MeOH); [α]26D −1469.4° (c 0.10, CHCl3), lit.1 [α]20D −1616.7° (c 0.1, CHCl3); 1H-NMR (CDCl3:CD3OD=1:1, TMS) δ 6.73 (1H, d, J=10.6 Hz) 6.43 (1H, dd, J=11.8, 14.4 Hz), 6.21 (1H, m), 6.21 (1H, m), 6.19 (1H, dd, J=10.9, 15.8 Hz), 6.18 (1H, d, J=15.4 Hz), 6.15 (2H, m), 6.10 (1H, m), 6.05 (1H, d, J=10.6 Hz), 6.02 (1H, dd, J=10.9, 14.4 Hz), 5.95 (1H, d, J=11.5 Hz), 5.65 (1H, d, J=15.8 Hz), 5.45 (1H, t, J=9.2), 5.45 (1H, m), 4.22 (1H, d, J=8.6 Hz), 4.17 (1H, m), 3.58 (3H, s), 2.28 (1H, m), 2.32 (1H, m), 2.09 (3H, s), 1.70 (3H, s), 1.66 (3H, s), 1.49 (3H, s), 1.32 (3H, d, J=6.7 Hz Hz); 13C-NMR (CDCl3:CD3OD=1:1) δ 167.0, 147.4, 143.4, 138.0, 137.8 × 3, 137.4, 135.5, 132.4 × 2, 130.7, 130.6, 130.0, 129.1, 128.1, 127.1, 125.4, 124.6, 76.2, 76.0, 61.3, 47.2, 38.5, 22.8, 20.9, 14.3, 13.0, 12.8; HRMS (ESI-TOF) m/z 466.2948 (466.2957 calcd for C29H40NO4, [M+H]+).
References
Futamura, Y. et al. Discovery of incednine as a potent modulator of the anti-apoptotic function of Bcl-xL from microbial origin. J. Am. Chem. Soc. 130, 1822–1823 (2008).
Tsujimoto, Y., Finger, L. R., Yunis, J., Nowell, P. C. & Croce, C. M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226, 1097–1099 (1984).
Reed, J. C., Cuddy, M., Slabiak, T., Croce, C. M. & Nowell, P. C. Oncogenic potential of bcl-2 demonstrated by gene transfer. Nature 336, 259–261 (1988).
Gross, A., McDonnell, J. M. & Korsmeyer, S. J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13, 1899–1911 (1999).
Ohtani, T. et al. Total synthesis of incednam, the aglycon of incednine. Org. Lett. 12, 5068–5071 (2010).
Grubbs, R. H. Olefin metathesis. Tetrahedron 60, 7117–7140 (2004).
Gradillas, A. & Pérez-Castells, J. Macrocyclization by ring-closing metathesis in the total synthesis of natural products: reaction conditions and limitations. Angew. Chem. Int. Ed. 45, 6086–6101 (2006).
Nicolaou, K. C., Bulger, P. G. & Sarlah, D. Metathesis reactions in total synthesis. Angew. Chem. Int. Ed. 44, 4490–4527 (2005).
Betzer, J. -F., Delaloge, F., Muller, B., Pancrazi, A. & Prunet, J. Radical hydrostannylation, Pd(0)-catalyzed hydrostannylation, stannylcupration of propargyl alcohols and enynols: regio- and stereoselectivities. J. Org. Chem. 62, 7768–7780 (1997).
Michels, T. D., Rhee, J. U. & Vanderwal, C. D. Synthesis of δ-tributylstannyl-α,β,γ,δ-unsaturated aldehydes from pyridines. Org. Lett. 10, 4787–4790 (2008).
Staudinger, H. & Meyer, J. New organic compounds of phosphorus. III. Phosphinemethylene derivatives and phosphinimines. Helv. Chim. Acta 2, 635–646 (1919).
Stuckwisch, C. G. Azomethine ylides, azomethine imines, and iminophosphoranes in organic syntheses. Synthesis 469–483 (1973).
Han, X., Stoltz, B. M. & Corey, E. J. Cuprous chloride accelerated Stille reactions. A general and effective coupling system for sterically congested substrates and for enantioselective synthesis. J. Am. Chem. Soc. 121, 7600–7605 (1999).
Schwab, P., France, M. B., Ziller, J. W. & Grubbs, R. H. A series of well-defined metathesis catalysts–synthesis of [RuCl2(:CHR’)(PR3)2] and its reactions. Angew. Chem. Int. Ed. 34, 2039–2041 (1995).
Scholl, M., Trnka, T. M., Morgan, J. P. & Grubbs, R. H. Increased ring closing metathesis activity of ruthenium-based olefin metathesis catalysts coordinated with imidazolin-2-ylidene ligands. Tetrahedron Lett. 40, 2247–2250 (1999).
Kingsbury, J. S., Harrity, J. P. A., Bonitatebus, P. J. Jr & Hoveyda, A. H. A recyclable Ru-based metathesis catalyst. J. Am. Chem. Soc 121, 791–799 (1999).
Garber, S. B., Kingsbury, J. S., Gray, B. L. & Hoveyda, A. H. Efficient and recyclable monomeric and dendritic Ru-based metathesis catalysts. J. Am. Chem. Soc. 122, 8168–8179 (2000).
Grela, K., Harutyunyan, S. & Michrowska, A. A highly efficient ruthenium catalyst for metathesis reactions. Angew. Chem. Int. Ed. 41, 4038–4040 (2002).
Bieniek, M., Michrowska, A., Gułajski, Ł. & Grela, K. A practical larger scale preparation of second-generation Hoveyda-type catalysts. Organometallics 26, 1096–1099 (2007).
Ohtani, T., Sakai, S., Takada, A., Takahashi, D. & Toshima, K. Efficient and stereoselective synthesis of the disaccharide fragment of incednine. Org. Lett. 13, 6126–6129 (2011).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Takada, A., Uda, K., Ohtani, T. et al. Improved total synthesis of incednam. J Antibiot 66, 155–159 (2013). https://doi.org/10.1038/ja.2013.4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2013.4
