
Abstract
Two new xanthone–anthraquinone heterodimers, acremoxanthone C (5) and acremoxanthone D (2), have been isolated from an extract of an unidentified fungus of the order Hypocreales (MSX 17022) by bioactivity-directed fractionation as part of a search for anticancer leads from filamentous fungi. Two known related compounds, acremonidin A (4) and acremonidin C (3) were also isolated, as was a known benzophenone, moniliphenone (1). The structures of these isolates were determined via extensive use of spectroscopic and spectrometric tools in conjunction with comparisons to the literature. All compounds (1–5) were evaluated against a suite of biological assays, including those for cytotoxicity, inhibition of the 20S proteasome, mitochondrial transmembrane potential and nuclear factor-κB.
Similar content being viewed by others

Introduction
Fungi have been a valuable source for drug leads.1, 2 As just one contemporary example that originated in The Journal of Antibiotics, the fungal metabolite, myriocin, initially called ISP-1 by Fujita et al. when first isolated from the entomopathogenic fungus Isaria sinclairii,3, 4 served as a lead-scaffold for the eventual development of fingolimod (FTY720), which received approval by the US FDA in 2010 for the treatment of multiple sclerosis.5 Despite numerous other drug leads from fungi, such as the often cited penicillin, it has been postulated that <10% of the ∼1.5 M fungi in the world have been investigated taxonomically.6 As part of a larger program to examine diverse natural product source materials, our team has been exploring solid phase cultures of filamentous fungi from the Mycosynthetix library, which has over 55 000 accessions, for new anticancer drug leads.1 In the course of this research, culture MSX 17022, which was isolated in April 1984 from leaf litter collected from a beech tree community near the Mycosynthetix headquarters in Hillsborough, NC, USA, yielded a cytotoxic extract, as evidenced by 93% inhibition of H460 cell growth when tested at 20 μg ml−1. Bioactivity-directed fractionation led to the isolation of a series of xanthone–anthraquinone heterodimers, two of which (compounds 2 and 5) were new. All compounds were examined against a suite of biological targets, including those for cytotoxicity, inhibition of the 20S proteasome, nuclear factor (NF)-κB inhibition assay and mitochondrial transmembrane potential activity.
Materials and Methods
General
Optical rotations, UV spectra and IR spectra were obtained on a Rudolph Research Autopol III polarimeter (Rudolph Research, Hackettstown, NJ, USA), a Varian Cary 100 Bio UV-vis spectrophotometer (Varian Inc., Walnut Creek, CA, USA) and a Perkin-Elmer Spectrum One with Universal ATR attachment (Perkin-Elmer, Waltham, MA, USA), respectively. NMR experiments were conducted in either dimethyl sulfoxide (DMSO)-d6 or CDCl3 with tetramethylsilane (TMS) as reference using a JEOL ECA-500 (operating at 500 MHz for 1H, 125 MHz for 13C; JEOL Ltd., Tokyo, Japan). High-resolution electrospray ionization mass spectrometry was performed on a Waters SYNAPT MS system (Waters Corp., Milford, MA, USA) using a capillary voltage of 2000 V (positive mode) and 1000 V (negative mode); sampling cone voltages were 40 V (positive mode) and 10 V (negative mode). Flash chromatography was conducted using a CombiFlash Rf system using a RediSep Rf Si-gel Gold column (both from Teledyne-Isco, Lincoln, NE, USA). HPLC was carried out on Varian Prostar HPLC systems (Walnut Creek, CA, USA) equipped with Prostar 210 pumps and a Prostar 335 photodiode array detector, with data collected and analyzed using Galaxie Chromatography Workstation software (version 1.9.3.2, Varian Inc.). For preparative HPLC, a Gemini-NX (5 μm; 250 × 21.2 mm) column was used with a 15 ml min−1 flow rate, whereas for analytical HPLC, a Gemini-NX (5 μm; 150 × 4.6 mm) column was used with a 1 ml min−1 flow rate (both from Phenomenex; Torrance, CA, USA).
Producing organism and fermentation
Mycosynthetix fungal strain 17022 was isolated by Dr Barry Katz of MYCOsearch in April of 1984 from leaf litter from a beech tree community in Hillsborough, NC, USA. DNA analyses were performed by MIDI Labs (Newark, DE, USA), and the D2 variable region of the large subunit rRNA was sequenced and compared with their database. This analysis suggested that this fungus was of the order Hypocreales; these data were deposited in Genbank (accession no. JN185925). The culture was stored on a malt extract slant and was transferred periodically. A fresh culture was grown on a similar slant, and a piece was transferred to a medium containing 2% soy peptone, 2% dextrose and 1% yeast extract (YESD media). Following incubation (7 days) at 22 °C with agitation, the culture was used to inoculate 50 ml of a rice medium, prepared using rice to which was added a vitamin solution and twice the volume of rice with H2O, in a 250 ml Erlenmeyer flask. This was incubated at 22 °C until the culture showed good growth (∼14 days). The scale-up culture was grown in a 2.8-l Fernbach flask (Corning, Inc., Corning, NY, USA) containing 150 g rice and 300 ml H2O and was inoculated using a seed culture grown in the YESD medium. This was incubated at 22 °C for 14 days.
Extraction and isolation
To the scale-up solid fermentation on rice was added 500 ml of 1:1 MeOH-CHCl3, and the mixture was shaken for 16 h on a reciprocating shaker. The solution was filtered and equal volumes of H2O and CHCl3 were added to the filtrate to bring the total volume to 2 l. The solution was stirred vigorously for 1 h, partitioned in a separatory funnel, and the bottom, organic layer was concentrated by rotary evaporation to dryness. This extract was stirred vigorously for 1 h in a mixture of 150 ml MeOH, 150 ml CH3CN and 200 ml hexane and then partitioned in a separatory funnel. The bottom layer was collected and evaporated to dryness (5.93 g). This defatted extract was adsorbed onto a minimal amount of Celite 545 (Acros Organics, Geel, Belgium) and dried with mixing via a mortar and pestle. This material was fractionated at 40 ml min−1 on a RediSep Rf Gold silica gel column (40 g, Teledyne-Isco), first with 100% hexanes for 0.7 column volumes (CV) followed by a gradient of 100% hexanes to 100% CHCl3 over 8.9 CV. The elution continued with 100% CHCl3 for 7.4 CV, then with a gradient of MeOH in CHCl3 (0–2% over 9.7 CV, then 2–5% over 5.2 CV, then 5–10% over 5.2 CV, then 10–20% over 3.7 CV, then 20–100% over 2.2 CV. MeOH (100%) was finally held for a further 6.7 CV). Fractions were collected every 24.75 ml. Compounds 1–4 were present in fractions 56–67, which were combined and evaporated (688.9 mg). Compound 5 was present in fractions 22–39 (370.9 mg). The combined fraction 56–67 was then subjected to preparative HPLC in three injections, 40–100% CH3CN in H2O over 30 min. Fractions were collected every 0.5 min. Elution times and amounts isolated were as follows: 1; 15–15.5 min (16.8 mg), 2; 19.5–20 min (8.4 mg), 3; 21–21.5 min (16.1 mg) and 4; 21.5–23.5 min (337.7 mg). The combined fraction 22–39 was purified by preparative HPLC using the identical conditions as above (in two injections), and compound 5 was collected between 26–28 min (327.5 mg).
Acremoxanthone C (5)
Compound 5 was isolated as a yellow solid (327.5 mg);. [α]D23 +576 (c 0.33, MeOH); UV (MeOH) λmax (log ɛ), 273 (4.03) and 371 (4.43) nm; IR (diamond) νmax 2956, 1736, 1634, 1566, 1469, 1368, 1219, 1097, 871 and 711 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Table 1 and Supplementary Information (Figures S3 and S4). HRMS m/z 637.1331 [M+Na]+; 613.1340 [M−H]− (calcd for C33H26O12Na, 637.1322; calcd for C33H25O12, 613.1346).
Acremoxanthone D (2)
Compound 2 was isolated as a yellow solid (8.4 mg);. [α]D23 +235 (c 0.33, MeOH); UV (MeOH) λmax (log ɛ), 287 (4.07) and 362 (4.17) nm; IR (diamond) νmax 3055, 1731, 1702, 1609, 1566, 1355, 1278, 1219, 1020 and 818 cm−1; 1H NMR (DMSO-d6, 700 MHz) and 13C NMR (DMSO-d6, 175 MHz), see Table 1 and Supplementary Information (Figures S1 and S2). HRMS m/z 653.1277 [M+Na]+; 629.1294 [M−H]− (calcd for C33H26O13Na, 653.1271; calcd for C33H25O13, 629.1295).
Conversion of 5 to 4
Compound 5 was stirred in pyridine at room temperature. The progress of the conversion of 5 to 4 was monitored by analytical RP-HPLC at 1, 2, 3 and 4.5 h.
Cytotoxicity assay
The cytotoxicity measurements against the MCF-77 human breast carcinoma (Barbara A Karmanos Cancer Center), NCI-H4608 human large cell lung carcinoma [HTB-177, American Type Culture Collection (ATCC)], SF-2689 human astrocytoma (NCI Developmental Therapeutics Program), HT-2910 human colorectal adenocarcinoma (HTB-38, ATCC) and the MDA-MB-43511 human melanoma (HTB-129, ATCC) cell lines were performed exactly as described in detail previously.12
20S proteasome assay
Human mammary adenocarcinoma cells7 (MCF-7; ATCC, Manassas, VA, USA) were propagated at 37 °C in 5% CO2 in RPMI 1640 medium supplemented with fetal bovine serum (10%) with penicillin (100 units ml−1) and streptomycin (100 μg ml−1). Cells in log phase growth were harvested by trypsinization followed by extensive washing to remove all traces of enzyme. A total of 7500 cells were seeded per well of a 96-well microtiter plate and incubated overnight (37 °C in 5% CO2). Samples dissolved in DMSO were then added to achieve the final concentrations as indicated (total volume: 100 μl; DMSO: 0.2%). The cells were incubated in the presence of test substance for 2 h at 37 °C and evaluated for proteasome activity using a commercial luminescent assay (Proteasome-Glo, Promega Corp., Madison, WI, USA) that measures the chymotrypsin-like, trypsin-like or caspase-like protease activity associated with the proteasome complex. Activity was expressed as percent inhibition relative to negative (solvent) control. The positive control was bortezomib tested at 5 nM and 25 nM, which inhibited proteasome activity by 27% and 91%, respectively.
NF-κB assay
An enzyme-linked immunosorbent assay based NF-κB inhibitory assay was performed exactly as described previously.12 Rocaglamide (Enzo Life Sciences International, Farmingdale, NY, USA) was used as a positive control (IC50 value of 0.075 μM).
Mitochondrial transmembrane potential (Δψ) assay
The mitochondrial transmembrane potential assay kit (Cayman Chemical Company, Ann Arbor, MI, USA) was adapted to detect the Δψ using a procedure published previously.13 Δψ is used to represent mitochondrial membrane transition events. The specific details were outlined recently,12 and staurosporine (Cayman) was used as a positive control (IC50 value of 2.5 nM).
Results and discussion
Major isolates
Identification of acremonidin A (4)
The numbering of the isolated compounds (1–5; see Figure 1) originates from their elution order on RP-HPLC (see Materials and Methods). However, the major compounds isolated in the course of this study were compounds 4 and 5, and thus, they will be discussed first. Compound 4 displayed a molecular formula of C33H26O12 from the HRMS data, indicative of an index of hydrogen deficiency of 21. From the diode array detector of the HPLC, the UV maxima were 285 and 359 nm. The Dictionary of Natural Products14 was utilized for dereplication, using the molecular formula and the UV maxima (with a range of ±10 nm from the observed maxima) as search criteria. The NMR data of 4 matched that of the single hit from Dictionary of Natural Products, which was acremonidin A,15 an antibacterial agent isolated from an Acremonium sp. in 2003.

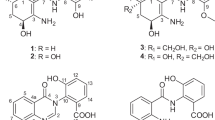
Structure of compounds (1-5) isolated from fungus MSX 17022.
Structure elucidation of acremoxanthone C (5)
The HRMS data of compound 5 yielded a molecular formula of C33H26O12, identical to that of 4. In the NMR spectra of compounds 4 and 5 (both examined in CDCl3), there was a high degree of similarity in those signals attributable to the anthraquinone portion of the molecule (for example, H-1′: δH 5.90 for 4, δH 5.96 for 5; H-3′: δH 6.82 for 4, δH 6.88 for 5; H-5′: δH 6.75 for 4 and δH 6.79 for 5). However, a key difference between the 1H NMR spectra of 4 and 5 was the number of phenolic peaks; 4 had six phenols whereas 5 only displayed four (Table 1). This could result from the formation of a xanthone in 5 via coupling of two of the phenols from rings E and F, with a resultant loss of water, as has been reported in the acremoxanthones.16 However, the formula for 5 did not allow for loss of water as compared with 4. The 13C NMR spectrum of 5 showed 33 resonances (Table 1), in agreement with the formula, 19 of which were quaternary, as opposed to 20 quaternary signals with 4. Moreover, the UV spectrum showed maxima at 274 and 369 nm, significantly different from that of 4.
Another ring-closing possibility, which had been reported previously, was via attack of one of the ring E phenols on the aromatic carbon bearing the methyl ester, as in the xanthoquinodins.17 2D-NMR data confirmed this type of ring-closure, and in this case, the linkage was through O-14. One new non-aromatic proton at δH 5.30 (H-3) and two new non-aromatic carbons at δC 74.8 (C-3) and 84.9 (C-2) were present in the NMR spectra of 5 as compared with 4. The signals for δH 5.30 (H-3) and δC 74.8 (C-3) correlated in the multiplicity-edited HSQC experiment, indicative of an aliphatic secondary hydroxyl group; this new H-3 signal correlated to H-4 and H-5 in the COSY spectrum as well. The δH 6.50 signal (H-4) correlated to the new quaternary carbon δC 84.9 (C-2) in the HMBC spectrum, whereas the H-5 signal (δH 6.13) correlated to the C-3 (δC 74.8) signal. The δH 6.50 resonance (H-4) also correlated to δC 171.2 resonance (C-6) in the HMBC spectrum, which was farther downfield than a typical phenol carbon, consistent with the enhanced keto character of C-6 resulting from tautomerization with the C-8 ketone. These data confirm the proposed E–G–F ring system. The relative stereochemistry of C-2 was confirmed by a 1D nOe difference spectrum, which showed an enhancement of H-3 on irradiation of the H3-16 methyl ester signal. The relative stereochemistry of C-1′, C-14′ and C-11′ was established by the ROESY spectrum. The acetate methyl group at C-18′ showed a clear ROESY correlation to H-13′, which established the relative stereochemistry for the anthraquinone portion of the molecule. Compound 5 therefore contained the anthraquinone portion of the acremonidins and acremoxanthones,15, 16 and the xanthone portion of the xanthoquinodins.17 This compound was named acremoxanthone C to maintain consistency with the existing acremoxanthones.
Minor isolates
Identification of moniliphenone (1)
According to HRMS data, compound 1 had the formula C16H14O6. The 1H NMR showed singlets due to aromatic methyl and methoxy groups. Aromatic region signals at δH 7.12 (doublet), 7.28 (triplet) and 7.47 (doublet), all with a coupling constant of 7.6 Hz, were consistent with a 1,2,3-trisubstituted benzene moiety. Using the AntiMarin database18 for dereplication, the formula was entered into the search query, as was one 1,2,3-trisubstituted benzene and one methoxyl. Only two hits resulted from this query; moniliphenone and nidulalin B19 both of which had 1H-NMR data that were consistent with the spectra of 1. However, the HMBC spectra revealed a key correlation between the δH 7.47 doublet and the δC 166.0 signal (ester carbonyl), establishing moniliphenone as the structure of 1; the 1H- and 13C-NMR data matched the literature.20
Structure elucidation of acremoxanthone D (2)
The HRMS data of compound 2 indicated a formula of C33H26O13, establishing an index of hydrogen deficiency of 21. The NMR data for compounds 2 and 5 were quite similar. The formula indicates the addition of one oxygen, similar to the difference of one oxygen between acremonidin A (4) and acremonidin C (3). Initially, it was expected that the extra oxygen of 2 would present as a hydroxy group at position C-9′, as was noted with acremonidin C (3).15 However, the HMBC spectrum did not confirm this. The oxygen instead was shown to be added as a hydroxy group at position C-7. Indeed, the C-7 resonance in the 13C-NMR spectrum of 2 was at δC 79.1, as opposed to δC 99.2 with 5. Correspondingly, a new singlet appeared in the 1H NMR spectrum of 2 at δH 8.13, with no HSQC correlation, indicating a hydroxy or phenol proton. This -OH proton correlated in the HMBC to the C-2 signal at δC 90.6, identifying the -OH as hydroxy. The carbon resonance for C-6 was farther downfield at δC 188.2 (vs 171.2 for 5), indicating more keto character due to the lack of tautomerization with C-8. The relative stereochemistry for 2 was established from the ROESY spectrum. The key observation was a strong correlation between the 7-OH proton and H-3. Of the four possible diastereomeric combinations of C-2 and C-7, only one possibility allows close approach of the 7-OH and H-3 according to Chem3D energy minimization, and that is with the C-2 methyl ester in the β position and the C-7 hydroxy in the α position. Because of the similarities between compounds 2 and 5, compound 2 was ascribed the trivial name, acremoxanthone D.
Identification of acremonidin C (3)
The HRMS of compound 3 indicated a formula of C33H26O13, identical to the formula for 2, however, the NMR spectra showed significant differences. From the diode array detector of the HPLC, the UV maxima were 275 and 341 nm, also differing significantly from 2 (287 and 362 nm). The Dictionary of Natural Products14 was again used for dereplication with molecular formula and UV maxima (with a range of ±10 nm from the observed maxima) as search criteria. A single hit was identified in Dictionary of Natural Products, and the NMR data of 3 matched that of acremonidin C.15
Conversion of compound 5 to 4
To determine the absolute configuration of compound 5, an attempt was made to synthesize the Mosher esters from the α-methoxy-α-trifluoromethylphenylacetyl chlorides (MTP chlorides) in pyridine.21 However, when the reaction was attempted, compound 4 was by far the major product, with numerous very minor products. When compound 5 was treated with pyridine only, the result was nearly complete conversion to 4 after 4.5 h at room temperature. This would indicate that H-3 of compound 5 is susceptible to basic attack followed by opening of the G-ring to yield 4 (Figure 2). This may not have been an issue with the xanthoquinodins (which also contain the E–G–F ring system), most likely because those compounds have a single bond between C-4 and C-5. The driving force behind the conversion of 5 to 4 may be the creation of aromaticity, which cannot occur with the xanthoquinodins. Therefore, absolute configurations were not determined for either 2 or 5.

Proposed conversion of compound 5 to 4 by pyridine at room temperature.
Biological activity
Compounds 1–5 were assayed against three cancer cell lines and showed moderate cytotoxic activity in vitro, with IC50 values summarized in Table 2. Compounds 2, 3 and 4 showed moderate 20S proteasome inhibitory activity as well (Table 2). All compounds were inactive in assays for both NF-κB inhibition and mitochondrial transmembrane potential (that is, IC50 values >20 μM; data not shown).
Conclusion
Using bioactivity-directed fractionation, five compounds were isolated from an extract of MSX 17022, an unidentified fungus of the order Hypocreales. Two of these compounds (2 and 5) were found to be new xanthone–anthraquinone heterodimers. The biological activity of these five compounds was found to be moderate for cytotoxicity and 20S proteasome inhibitory activity (Table 2). Regardless, the biosynthetic potential of this fungus was quite intriguing, particularly the generation of xanthone–anthraquinone heterodimers. Even a non-optimized culture, first isolated nearly three decades ago, produced structurally diverse compounds on the scale of hundreds of mg per 2.8 l culture.
Accession codes
References
Orjala, J., Oberlies, N. H., Pearce, C. J., Swanson, S. M. & Kinghorn, A. D. Discovery of potential anticancer agents from aquatic cyanobacteria, filamentous fungi, and tropical plants. in Bioactive Compounds from Natural Sources. Natural Products as Lead Compounds in Drug Discovery (ed. Tringali, C.) 37–63 2nd edn Taylor & Francis, London, UK, 2012.
Pearce, C., Eckard, P., Gruen-Wollny, I. & Hanske, F. G. Microorganisms: their role in the discovery and development of medicines. in Natural Product Chemistry for Drug Discovery (eds A. D. Buss & M. S. Butler) 215–244 The Royal Society of Chemistry, Cambridge, UK, 2010.
Fujita, T. et al. Fungal metabolites. Part 11. A potent immunosuppressive activity found in Isaria sinclairii metabolite. J. Antibiot. 47, 208–215 (1994).
Fujita, T. et al. Fungal metabolites. Part 12. Potent immunosuppressant, 14-deoxomyriocin, (2S,3R,4R)-(E)-2-amino-3,4-dihydroxy-2-hydroxymethyleicos-6-enoic acid and structure-activity relationships of myriocin derivatives. J. Antibiot. 47, 216–224 (1994).
Strader, C. R., Pearce, C. J. & Oberlies, N. H. Fingolimod (FTY720): a recently approved multiple sclerosis drug based on a fungal secondary metabolite. J. Nat. Prod. 74, 900–907 (2011).
Hawksworth, D. L. & Rossman, A. Y. Where are all the undescribed fungi? Phytopathology 87, 888–891 (1997).
Soule, H. D., Vazguez, J., Long, A., Albert, S. & Brennan, M. A human cell line from a pleural effusion derived from a breast carcinoma. J. Natl. Cancer Inst. 51, 1409–1416 (1973).
Carney, D. N., Gazdar, A. F., Bunn, P. A. Jr. & Guccion, J. G. Demonstration of the stem cell nature of clonogenic tumor cells from lung cancer patients. Stem Cells 1, 149–164 (1982).
Rosenblum, M. L. et al. Stem cell studies of human malignant brain tumors. Part 1: development of the stem cell assay and its potential. J. Neurosurg. 58, 170–176 (1983).
Fogh, J. & Trempe, G. New human tumor cell lines. in Human Tumor Cells In Vitro 115–159 Plenum, New York, USA, 1975.
Rae, J. M., Creighton, C. J., Meck, J. M., Haddad, B. R. & Johnson, M. D. MDA-MB-435 cells are derived from M14 melanoma cells—a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res. Treat. 104, 13–19 (2007).
Ayers, S. et al. Resorcylic acid lactones with cytotoxic and NF-κB inhibitory activities and their structure-activity relationships. J. Nat. Prod. 74, 1126–1131 (2011).
Deng, Y. et al. Bioactive 5,6-dihydro-α-pyrone derivatives from Hyptis brevipes. J. Nat. Prod. 72, 1165–1169 (2009).
Dictionary of Natural Products. Chapman & Hall/CRC Chemical Database, from http://www.chemnetbase.com.
He, H., Bigelis, R., Solum, E. H., Greenstein, M. & Carter, G. T. Acremonidins, new polyketide-derived antibiotics produced by Acremonium sp., LL-Cyan 416. J. Antibiot 56, 923–930 (2003).
Isaka, M., Palasarn, S., Auncharoen, P., Kornmijit, S. & Jones, E. B. G. Acremoxanthones A and B, novel antibiotic polyketides from the fungus Acremonium sp. BCC 31806. Tetrahedron Lett. 50, 284–287 (2009).
Tabata, N., Tomoda, H., Matsuzaki, K. & Omura, S. Structure and biosynthesis of xanthoquinodins, anticoccidial antibiotics. J. Am. Chem. Soc. 115, 8558–8564 (1993).
AntiMarin Database, Blunt, J. W., Munro, M. H. G. & Laatsch, H. Eds.;University of Canterbury, Christchurch, New Zealand; and University of Göttingen, Göttingen, Germany, (2010).
Kawahara, N., Sekita, S., Satake, M., Udagawa, S. & Kawai, K. Structures of a new dihydroxanthone derivate, nidulalin A, and a new benzophenone derivative, nidulalin B, from Emericella nidulans. Chem. Pharm. Bull. 42, 1720–1723 (1994).
Kachi, H. & Sassa, T. Isolation of monilphenone, a key intermediate in xanthone biosynthesis from Monilinia fructicola. Agric. Biol. Chem. 50, 1669–1671 (1986).
Hoye, T. R., Jeffrey, C. S. & Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2, 2451–2458 (2007).
Acknowledgements
This research was supported by P01 CA125066 from the National Cancer Institute/National Institutes of Health, Bethesda, MD, USA. The Golden LEAF Foundation (Rocky Mount, NC, USA) provided partial support to DJK. Mycology technical support was provided by Maurica Lawrence. The authors thank Mingming Su of the David H Murdock Research Institute, Kannapolis, NC, USA for high-resolution mass spectrometry data.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article
Cite this article
Ayers, S., Graf, T., Adcock, A. et al. Cytotoxic xanthone–anthraquinone heterodimers from an unidentified fungus of the order Hypocreales (MSX 17022). J Antibiot 65, 3–8 (2012). https://doi.org/10.1038/ja.2011.95
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2011.95
Keywords
This article is cited by
-
Revisiting the enniatins: a review of their isolation, biosynthesis, structure determination and biological activities
The Journal of Antibiotics (2012)
