
Abstract
In the course of our chemical and biological screening program for yet unidentified microbial metabolites, we selected plants of Traditional Chinese Medicine (TCM) as habitats for talented Streptomycetes producer strains for the first time. Liquid pure cultures of strain Streptomyces sp. GS DV232 were found to contain 4-methyl-2-quinazolinamine (1), a potent alkaloid yet unknown from nature. In this study, we investigated the chemical and crystal structure of 1, as well as its antiproliferative bioactivity, and addressed the unusual biosynthesis using feeding experiments.
Similar content being viewed by others
Introduction
On the basis of knowledge of traditional medicine, natural products as extracts or pure compounds make a crucial contribution to the health precautions of about 80% of the world's population today.1 In addition to plants being the main category, selective groups of microorganisms such as actinomycetes, myxobacteria and various fungi are called ‘talented producers’ of secondary metabolites. They provide important drugs for clinical use, such as antibiotics, antitumor agents or especially immunosuppressive compounds; in these fields of applications, natural products are by far unmatched by synthetic chemicals. Furthermore, microbial metabolites are a source of research biochemicals and agrochemicals and are continuously investigated worldwide. To a significant extent, expertise in modern organic chemistry in terms of structural diversity, reaction types and mechanisms is driven by exploring natural products.2, 3, 4 In addition to the indispensable use for medicinal and agricultural applications, the innovative potential of natural products lies in the discovery of manifold ecological interactions, which are presently unknown. These are explored in interdisciplinary research networks of biology, ecology and chemistry.5 Screening for new metabolite structures by the ‘ethnobotanical approach’ takes advantage of the tradition of using medicinal plants by indigenous people.6, 7 Ethnobotany is considered as the science of transferring knowledge of historical therapy into active principles of drug agents. Thus, drug discovery benefits from rare or yet unexplored habitats, reinforcing this research strategy.1, 8
Our ongoing screening project for yet unknown microbial metabolites comprises the isolation of microorganisms from Traditional Chinese Medicinal (TCM) herbs with the aim of isolating strains, establishing laboratory cultivations and giving access to bioactive substances from a thus renewable resource. Among 51 bacterial isolates, Streptomyces sp. GS DV232 was isolated from leaves (folium) of Filipendula palmata (Pall.) Maxim. (bot. syn.: Spiraea palmata Pall., Chinese: wen zi cao). Traditionally, the dried rhizome or herb is used to treat the respiratory system and is also used against rheumatic and renal diseases.9, 10 From strain GS DV232, quinazolinamine 1 could be isolated as the main secondary metabolite.

Hitherto, the structurally uncommon 1 has not been found in nature, and has only been described as a synthetic chemical product.11, 12 Among plant alkaloids, small quinazolines are well known. However, only a few single reports deal with quinazolines as secondary metabolites of microorganisms.13, 14 In this study, cultivation, isolation, structural elucidation, crystal structure and biological activity of 1 are presented. Feeding experiments allow for insights into the unusual biosynthetic pathway of microbial quinazoline 1.
Results and discussion
Isolation and structural elucidation
Streptomyces sp. GS DV232 was isolated from leaf surfaces of traditionally used plants of the Filipendula palmata (Pall.) Maxim. Owing to 16S rDNA analysis, it could be taxonomically classified as Streptomyces zaomyceticus or S. venezuelae. According to the chemical screening strategy, strain GS DV232 was cultivated in different media to analyze its metabolite pattern. The obtained extracts showed several metabolites using high performance thin layer chromatography, but only one gave an intense red color reaction on TLC plates after staining with anisaldehyde. Chromatography gave a white solid with an overall yield of 2 mg l−1, the molecular formula (C9H9N3) of which was determined by mass spectral analysis (HRESI mass spectrometry, [M+H]+ 160.08692). 1H-NMR spectra exhibited four aromatic protons, a methyl group (2.72 p.p.m.) with distinct NOESY correlations to the aromatic proton at 7.94 p.p.m. and a broad singlet at 6.08 p.p.m. for an amino group. From 13C-NMR, HSQC and HMBC data, the compound was identified as the alkaloid 4-methyl-2-quinazolinamine (1). Hitherto, 1 has not been described from any natural source. Given that the number of synthetic quinazoline derivatives exceeds the number of ‘natural’ quinazolines,13 1 was first synthesized by Theiling and McKee12 during their studies on heterocyclic compounds carrying guanidine substituents.
For further investigations, 1 was synthesized according to Hynes et al.11 and physicochemical properties were verified. Crystals suitable for X-ray structure determination were obtained from a CD2Cl2 solution of the synthetic material by evaporation. The crystal structure underlined the formula of 1 (Figure 1).15 Nitrogen N-10 was found to bind two hydrogen atoms, the positions of which were clearly visible in the electron density. Although potential hydrogen positions at N-1 and N-3 also showed a slight residual density, the crystal structure is mainly based on amine 1. The crystal lattice is based on hydrogen bonds between the amino hydrogen atoms and the free electron pairs at N-1 and N-3, as shown in Figure 1. At present, only one related crystal structure can be found in the CSD (Cambridge Structural Database), which resembles [1,2,4]triazoloquinazolinium betaine (2).16

Structure 1 as determined by X-ray crystallographic analysis.15 The atoms are shown as ORTEP plots with 50% probability displacement ellipsoids. Dashed lines mark hydrogen bonds.
Feeding experiments
So far, only very few reports have addressed the biosynthesis of the small bicyclic quinazolines. Among the known quinazoline alkaloids from plants, arborine (3) and peganine/vasicine (4) were examined by in vivo feeding experiments.17, 18, 19 Anthranilic acid has been suggested to be the putative precursor of the aromatic ring system and phenylalanine for the pyrimidine fragment. Yet, the metabolic origin of this aminobenzoic acid has only been discussed. Incorporation of labeled tryptophan [U-14C6] benzene ring into 4 fueled questions whether anthranilic acid derives de novo from the shikimate pathway or from the catabolism of tryptophan.19, 20 4-Methylquinazolines from Pseudomonas sp. have been suggested to originate from the so-called ‘quinazoline pathway’, starting from tryptophan through N-formylacetophenone to yield 4-methylquinazolines of type 5.21 Therefore, we are fascinated to have a bacterial 4-methyl-2-quinazolinamine (1) at hand and to investigate the biosynthesis that presumably encloses anthranilic acid, some urea-like nitrogen source and possibly methionine for the C-4 methyl group. Our feeding experiments with unlabeled anthranilic acid, glycerol and urea should show whether these compounds increased the production of 1. No alteration for quinazoline (1) production has been detected by TLC and HPLC-MS analysis.

Shikimate generally incorporates glycerol from phosphoenolpyruvate (PEP) and erythrose-4-phosphate (E4P) of the pentose phosphate pathway and gives rise to specifically labeled anthranilic acid (Figure 2). Therefore, 13C3-labeled glycerol was administered to growing cultures, and 13C-NMR spectra of purified 1 clearly showed a distinct 13C-13C coupling between C-5/C-6/C-7 and C-4a/C-8a and a single enrichment at methyl group C-9 (Supplementary information). This indicates an intact incorporation of glycerol through the shikimate pathway (Figure 2). Levels of [13C3]glycerol are unbalanced in the metabolic pools of PEP and E4P, and therefore C-8 (C-1 of E4P) might not be significantly labeled.22

Labeling patterns of (a) shikimate from glycerol metabolism in general; and (b) quinazoline 1 from [U-13C3]glycerol and [methyl-13C]methionine.
Previous speculations about the incorporation of methionine into quinazoline alkaloids revolve around the origin of the methyl group at N-1 of 3. Gröger and collaborators17 isolated labeled N-methylanthranilic acid as a degradation product during their studies. [Methyl-13C]methionine did not significantly label any carbon atom of 1, and N-methylanthranilic acid has not been isolated from feeding studies with strain GS DV232 (data not shown), but in earlier experiments. Uncommonly, 4-methyl-2-quinazolinamine (1) bears a third nitrogen atom in the pyrimidine fragment, which strongly invited for biosynthetic experiments. [13C]Urea was fed to the producing strain and gave no significant enrichments. However, consistent labeling patterns for 1 were observed from feeding [15N2]urea and [13C,15N2]urea. The 1H-NMR spectra revealed new high field-shifted multiplet signals, δH=6.07 p.p.m. (1JNH 88.5 Hz, 3JNH 5.5 Hz), pointing to an incorporation rate of about 14% of 15N into the NH2-group. 1H-15N-HMBC studies (with optimization on JNH 5 Hz) gave only a significant cross-correlation of N-3 (δN= −110 p.p.m.) and 9-CH3 (δH=2.72 p.p.m.). In addition, several JNC couplings are deduced from 13C-NMR data (Figure 3), suggesting a distinguished, moderate 15N incorporation into all N-atoms of 1 because of a vital catabolism of 15N-labeled urea, but no incorporation of an intact [13C,15N]urea moiety.

Coupling constants (in Hz) of 1 from 1H-NMR (JNH: gray) and 13C-NMR data (JNC: black) after feeding [13C,15N2]urea.
From our data of the comprehensive feeding experiments, we suggest the following biosynthetic pathway for the uncommon microbial 2-quinazolinamine metabolite 1:
-
1)
Tryptophan originates from anthranilic acid from the shikimic acid pathway by means of PEP and E4P. Three sources form the tryptophan skeleton, anthranilate (shikimic acid pathway), 5-phosphoribosylpyrophosphate (pentose phosphate pathway) and serine (3-phosphoglycerate from glycolysis), which in turn are formed from glycerol (Figure 4).23
Figure 4 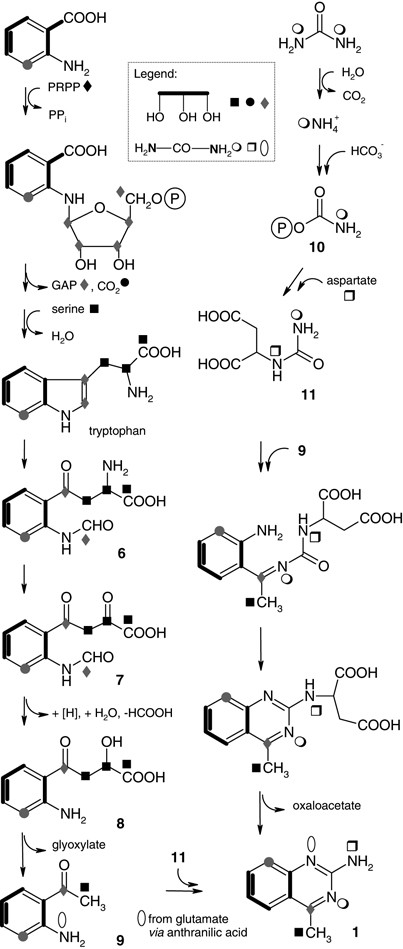
The proposed biosynthetic pathway for 4-methyl-2-quinazolinamine (1) in accordance with labeling patterns derived from 13C- and 15N-labeled precursors (black: significant, grey: weak/not detectable incorporation).
-
2)
Cleavage of tryptophan by tryptophan pyrrolase/tryptophan-2,3-dioxygenase could form N-formylkynurenine (6), which is transformed into 7 by a 2-oxoglutarate aminotransferase. Possibly, a reduction at the 2-keto group gives 8, which is converted into 2-aminoacetophenone (9) and glyoxylate through a ketohydroxyglutarate aldolase reaction type (Figure 4).24
-
3)
The significant high incorporation of labeled urea in NH2-10 and the indicated weak labeling of N-1 and N-3 should be the result of urea catabolism by urease—already known from Streptomyces—to NH3 and CO2. The resulting 15N-labeled ammonium represents a source for each of the three nitrogens in 1 in individual degrees. Hence, no [13C]label is transferred directly from [13C,15N]urea into carbon C-2 of 1. That is, N-3–C-2–N-10 does not come from an intact urea-moiety (Figure 4).
-
4)
We suggest that inorganic ammonium labels carbamoyl phosphate (10), which mediates 15N transfer to 1. We consequently assume N-carbamoyl aspartate (11) carrying 15N isotopic labels derived from 10 and from glutamate in the course of aspartate biosynthesis by transamination. Generally, glutamate in turn is biosynthetically close to the ammonia pool, and is therefore strongly 15N labeled in respective feeding experiments.
-
5)
It is also feasible that N-1 is 15N labeled, as glutamate also provides the anthranilic acid and indole nitrogen at an early stage of biosynthesis (Figure 4). To conclude, labeling of the three nitrogen atoms in different intensities reflects the dynamic metabolism (time, sites, substrate specificities).
-
6)
Finally, the quinazoline ring system is formed from 9 and 11 by a double nucleophilic attack (Figure 4). Transamination and release of oxaloacetate could result in the new quinazoline metabolite 1.

Biological activity
For biological screening, crude extracts of strain GS DV232 were tested against different test organisms in a plate diffusion assay. A weak growth inhibition of Bacillus subtilis and Escherichia coli was observed, but no growth inhibition of any testorganisms could be detected for the pure 4-methyl-2-quinazolinamine (1). Cytotoxicity cell assays with 1 indicated a moderate activity against different tumor cell lines (Table 1). Furthermore, 1 showed antiproliferating properties on bronchial carcinoma cells (line A549), which can be compared with known chemotherapeutic agents such as doxorubicin and melphalan (Figure 5) (Supplementary information). Currently, ongoing assays are concerned with the possible mode of action of 1, and thus the morphological effects on tumor cells were investigated. After incubating breast adenocarcinoma cells (line MCF 7) with 1 in a concentration of 5 μg ml−1, the cells showed a deformation of the nuclei combined with hollows in the network of microtubules (Supplementary information).

Antiproliferative potential of 1 against bronchial carcinoma cells (line A549) compared with doxorubicin and melphalan (Supplementary information). Relative colony-forming rates (%) were obtained as follows: average of treated colonies/average of untreated colonies × 100.
Conclusion
The concept of our screening has its seeds in TCM plants as habitats for potent antibiotic-producing Streptomycetes, one of them producing 4-methyl-2-quinazolinamine (1), not known as a natural product before. Significant enrichments of 2-quinazolinamine 1 from feeding experiments indicated the biosynthetic pathway of 1 to originate from a tryptophan-derived core. The three nitrogen atoms are an uncommon feature of the bacterial quinazoline 1; in this study, we present a conclusive biosynthetic pathway. In different biological tests for antitumor activity, 1 showed a moderate antiproliferating potential on cancer cells in comparison with known compounds. Thus, ongoing research revolves around the synthesis and biological evaluation of 2-amino-substituted quinazolines.
Although alkaloids—mainly of plant origin—have been intensively investigated, quinazolines especially are still in the focus of research. Streptomycetes have been observed to produce known alkaloids and the strikingly simple quinazoline-4-one.25 We learn that (synthetically) known quinazoline structures have been also invented by nature: 4-methylquinazoline (5a), first discovered from Pseudomonas sp.,21 was recently identified as a sex pheromone in the parasitoid wasp Nasonia vitripennis.26 Synthetic efforts on 2-guanidino quinazolines as adenosine receptor antagonists add on the picture of actual quinazoline research fields.27
Experimental section
General
Electron impact and HREI mass spectra were recorded on Finnigan MAT 95 (70 eV; Thermo Scientific, Dreieich, Germany); relative intensities in parenthesis refer to the highest peak of the spectrum. HRESI-MS spectra were obtained using Bruker Apex IV (FTICR, 7 T; Bruker Daltonik GmbH, Bremen, Germany). All NMR spectra were measured in acetone-d6 or CD2Cl2, with the solvent as internal reference. Chemical shifts are expressed in δ-values. 1H-NMR spectra were recorded on Varian Inova-600, and 13C-NMR spectra on Varian Inova-500 (125.7 MHz) equipped with Varian PFG Cold Probe (with helium cooling system). 15N-NMR spectra were recorded on Varian VNMR-S 600 equipped with a 3 mm triple-resonance inverse probehead (temperature 25 °C, 600 MHz for 1H and 60 MHz for 15N). UV spectra were obtained in methanol from a Varian Cary 3E spectrometer (Varian GmbH, Darmstadt, Germany), and IR spectra from a PerkinElmer model 1600 as KBr pellets (PerkinElmer LAS GmbH, Rodgau, Germany). M.p.s were measured with a Reichert (Vienna, Austria) hot-stage microscope (not corrected).
Producer strain
The producing strain Streptomyces sp. GS DV232 was isolated from the leaf (folium) of the meadowsweet Filipendula palmata (Pall.) Maxim. family of Rosaceae, which was collected near Harbin, Heilongjiang Province, PR of China in 2005. Taxonomic investigations suggest strain GS DV232 to be S. zaomyceticus and S. venezuelae (100% identity by 16S rDNA analysis).
Chemical screening and isolation
Chemical screening was performed by the cultivation of strain GS DV232 in different media with oat meal (HF), soybean meal (SGG) and yeast/malt/glucose (YMG), and subsequent chemical and biological analyses of the extracts (Supplementary information). Fermentations were carried out in liquid cultures (1 l baffled Erlenmeyer flasks with 250 ml broth volumes) that were incubated in a shake incubator at 28 °C, 180 r.p.m. for 72 h. After adjusting the pH to 5.0, culture broths were filtrated. Filtrates were extracted thrice with ethyl acetate, and combined organic phases were evaporated. Crude extracts were subjected to chromatography on silica gel (eluent: chloroform/methanol 9:1), followed by MPLC (medium pressure liquid chromatography) on RP-18 (Merck LiChroprep column; Merck KGaA, Darmstadt, Germany; eluent: methanol/water 8:2). Optional further purification was achieved by HPLC (high performance liquid chromatography): Jasco PU-2080plus pump, Jasco MD-210plus UV-detector, Rheodyne manual injection valve, software Jasco Borwin (version 1.50) with Jasco HSS-2000 (version 3.5.2) (Jasco GmbH, Gross-Umstadt, Germany); column: Phenomenex Aqua (Hydro-RP; Phenomenex, Aschaffenburg, Germany) C18 endc., 5 μm, 250 × 10 mm, flow rate 2.7 ml min−1; isocratic solvent system: MeOH/H2O 15:85; UV detection at 274 and 339 nm; retention time of 7 min for 1. The overall yield of 1 was 2 mg l−1 from liquid cultures.
Feeding experiments
For feeding experiments, the producer strain was cultivated in a 2 l fermenter (Biostat B; B.Braun AG, Melsungen, Germany) containing 1.5 l YMG medium for 54 h. A 48 h preculture in 1 l baffled Erlenmeyer flasks with 150 ml YMG medium was used as inoculum. From the twelfth to the twenty-third hour of fermentation, feeding reagents were added continuously as sterile-filtered solutions (40 ml each). Feeding reagents: anthranilic acid 7.3 mM, glycerol 5.4 mM, [U-13C3]glycerol (99% 13C) 5.4 mM, urea 4.0 mM, [13C]urea (99% 13C) 4.0 mM, [15N2]urea (95% 15N) 4.0 mM, [13C,15N2]urea (99% 13C, >98% 15N) 4.0 mM and [methyl-13C]methionine (99% 13C) 1.2 mM. Labeled glycerol, [13C]urea and [15N2]urea were obtained from Campro Scientific GmbH (Berlin, Germany), and [13C,15N2]urea and methionine from Cambridge Isotope Laboratories (Euriso-top GmbH, Saarbrücken, Germany). Isolation of pure 1 was performed by subjecting the crude extract to chromatography on Sephadex LH-20 (Sigma-Aldrich Chemie GmbH, Munich, Germany; eluent: methanol), followed by MPLC and HPLC as described above.
Biological tests
In plate diffusion tests, crude extracts and pure compounds were tested against B. subtilis, Candida albicans, E. coli and Staphylococcus aureus (Supplementary information).
In a first test series, cytotoxic activity was determined according to NCI (USA) protocol with tumor cell lines AGS, HM02 (gastric adenocarcinoma), HepG2 (hepatocellular carcinoma) and MCF 7 (breast adenocarcinoma) (Supplementary information).28 In the second study, 1 was tested with regard to its toxic potential on the colony-forming rate of human bronchial carcinoma cells (line A549) (Supplementary information). For morphological studies, MCF 7 (breast adenocarcinoma) cells were incubated for 24 h after adding 1 in a concentration of 5 μg ml−1. Microtubules were colored green by immunofluorescence, nuclei and chromosomes were stained blue with 4,6-diamidino-2-phenylindole (Supplementary information).
Crystal structure determination
1 was crystallized in a Ø 3 mm glass vial by evaporation from CD2Cl2. A suitable crystal (0.27 × 0.22 × 0.18 cm) was mounted at 100 K on a Bruker Smart 6000 CCD diffractometer (Bruker AXS GmbH, Karlsruhe, Germany) equipped with mirror-system-monochromated Cu Kα radiation (λ 1.54178 Å). A total of 8189 reflections were measured, of which 1197 were independent. Data reduction and processing was carried out with SAINT (Bruker AXS), empirical absorption correction with SADABS (Bruker AXS) and phasing with SHELXS.29 The asymmetric unit contained two molecules and the measured crystal belonged to the orthorhombic space group P 21 21 21. The structure was determined by direct methods. All atoms except hydrogens were refined anisotropically by the full-matrix least-squares method on F2 using SHELXL9729 to give a final R-factor of 0.0226. The amino hydrogen atoms were refined, all other hydrogen atoms were calculated in riding positions.
4-Methyl-2-quinazolinamine (1)
C9H9N3 (159.19);. white solid; m.p. 112 °C; Rf 0.47 (RP-18, MeOH/H2O 8:2); EI-MS (70 eV) m/z (%) 159 (100) [M]+, 144 (12) [M-CH3]+, 132 (18), 119 (17); HREI-MS calcd., found m/z 159.0797; HRESI-MS calcd., found m/z 160.08692 [M+H]+; UV (MeOH) λmax nm (log ɛ) 202 (4.04), 235 (4.28), 349 (3.31); UV (MeOH+HCl) λmax nm (log ɛ) 201 (3.96), 230 (4.14), 246 (4.01), 275 (3.43), 342 (3.34); UV (MeOH+NaOH) λmax nm (log ɛ) 211 (3.81), 236 (4.31), 350 (3.57); IR νmax (KBr) cm−1 3329, 3176, 1650, 1617, 1565, 1480, 1441, 1392, 1363, 1263, 1142, 1037; 1H-NMR (600 MHz, acetone-d6): δ p.p.m. 2.72 (s, 3H, 9-H3), 6.08 (bs, 2H, 2-NH2), 7.21 (ddd, J=8.5, 7.0, 1.5 Hz, 1H, 6-H), 7.44 (dd, J=8.5, 1.5 Hz, 1H, 8-H), 7.63 (ddd, J=8.5, 7.0, 1.5 Hz, 1H, 7-H), 7.94 (dd, J=8.5, 1.5 Hz, 1H, 5-H); 1H-NMR (600 MHz, CD2Cl2) (see Supplementary information); 13C-NMR (125.7 MHz, acetone-d6) δ p.p.m. 21.5 (q, C-9), 120.3 (s, C-4a), 122.9 (d, C-6), 126.4 (d, C-5), 126.7 (d, C-8), 134.3 (d, C-7), 153.2 (s, C-8a), 161.4 (s, C-2), 170.6 (s, C-4); 13C-NMR (125.7 MHz, CD2Cl2) (see Supplementary information); 15N-NMR (60 MHz, acetone-d6) δ p.p.m. −110.4 (N-3), −164.2 (N-1), −303.1 (N-10). Chemical shifts were obtained from 1H-15N-HMQC resp. 1H-15N-HMBC correlations. Chemical synthesis of 1 was performed considering the results of Hynes et al.11 (Supplementary information).
References
Gurib-Fakim, A. Medicinal plants: traditions of yesterday and drugs of tomorrow. Mol. Aspects Med. 27, 1–93 (2006).
Newman, D. J. & Cragg, G. M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 70, 461–477 (2007).
Cordell, G. A. Natural products in drug discovery—creating a new vision. Phytochem. Rev. 1, 261–273 (2002).
Wilson, R. M. & Danishefsky, S. J. Small molecule natural products in the discovery of therapeutic agents: the synthesis connection. J. Org. Chem. 71, 8329–8351 (2006).
Lam, K. S. New aspects of natural products in drug discovery. Trends Microbiol. 15, 279–289 (2007).
Heinrich, M. Ethnobotany and natural products. Curr. Top. Med. Chem. 3, 141–154 (2003).
Strobel, S. A. & Strobel, G. A. Plant endophytes as a platform for discovery-based undergraduate science education. Nat. Chem. Biol. 3, 356–359 (2007); and citations therein.
Izzo, A. A. & Ernst, E. Interactions between herbal medicines and prescribed drugs: a systematic review. Drugs 61, 2163–2175 (2001).
Traversier, R., Staudinger, K. & Friedrich, S. TCM in westlichen Pflanzen, Phytotherapie, Akupunktur, Diätetik. Sonntag Verlag Stuttgart (2005) (in German).
Wu, Z. Y., Raven, P. H. & Hong, D. Y. (eds). Flora of China Vol. 9 (Pittosporaceae through Connaraceae). (Science Press, Beijing, China and Missouri Botanical Garden Press, St Louis, MO, USA, 2003).
Hynes, J. B., Campbell, J. P. & Hynes, J. D. Synthesis of 2-aminoquinazolines from ortho-fluoroketones. J. Heterocyclic Chem. 32, 1185–1187 (1995).
Theiling, L. F. & McKee, R. L. 2-Guanidinoquinazolines. J. Am. Chem. Soc. 74, 1834–1836 (1952).
D’yakonov, A. L. & Telezhenetskaya, M. V. Quinazoline alkaloids in nature. Chem. Nat. Compd. 33, 221–267 (1997).
Michael, J. P. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 25, 166–187 (2008).
CCDC reference number 737855 contains the supplementary crystallographic data for this structure. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033; e-mail: deposit@ccdc.cam.sc.uk).
Crabb, D. L. et al. Preparation of [1,2,4]triazoloquinazolinium betaines and molecular rearrangements of putative [1,2,4]triazolo[4,3-a][1,3,5]triazinium betaines. J. Chem. Soc., Perkin Trans. 1, 1517–1525 (1999).
Johne, S., Waiblinger, K. & Gröger, D. Zur Biosynthese des Chinazolinalkaloids Arborin. Eur. J. Biochem. 15, 415–420 (1970) (in German).
O’Donovan, D. G. & Horan, H. The biosynthesis of arborine. J. Chem. Soc. (C) 1970, 2466–2470 (1970).
Liljegren, D. R. Biosynthesis of quinazoline alkaloids of Peganum harmala. Phytochemistry 10, 2661–2669 (1971).
Liljegren, D. R. The biosynthesis of quinazoline alkaloids of Peganum harmala L. Phytochemistry 7, 1299–1306 (1968).
Mann, S. Chinazolinderivate bei Pseudomonaden. Arch. Mikrobiol. 56, 324–329 (1967) (in German).
Wenzel, S. C. et al. On the biosynthetic origin of methoxymalonyl-acyl carrier protein, the substrate for incorporation of ‘glycolate’ units into ansamitocin and soraphen. J. Am. Chem. Soc. 128, 14325–14336 (2006).
Berg, J. M., Tymoczko, J. L. & Stryer, L. Biochemistry, International 6th edition. (W.H. Freeman & Co Ltd, New York, USA, 2006).
Nishihara, H. & Dekker, E. E. Purification, substrate specifity and binding, β-decarboxylase activity, and other properties of Escherichia coli 2-keto-4-hydroxyglutarate aldolase. J. Biol. Chem. 241, 5079–5087 (1972).
Maskey, R. P., Shaaban, M., Grün-Wollny, I. & Laatsch, H. Quinazoline-4-one derivatives from Steptomyces isolates. J. Nat. Prod. 67, 1131–1134 (2004).
Ruther, J., Steiner, S. & Garbe, L.-A. 4-Methylquinazoline is a minor compound of the male sex pheromone in Nasonia vitripennis. J. Chem. Ecol. 34, 99–102 (2008).
Webb, T. R. et al. Quinazolines as adenosine receptor antagonists: SAR and selectivity for A2B receptors. Bioorg. Med. Chem. 11, 77–85 (2003).
Grever, M. R., Schepartz, S. A. & Chabner, B. A. The National Cancer Institute: cancer drug discovery and development program. Semin. Oncol. 19, 622–638 (1992).
Sheldrick, G. M. A short history of SHELX. Acta. Cryst. A64, 112–122 (2008).
Acknowledgements
We thank Prof Dr W Beil (Institute of Pharmacology, Medizinische Hochschule Hannover/Germany) and Dr F Sasse (Helmholtz Centre for Infection Research, Braunschweig/Germany) for performing cell assays. We thank Prof Dr Fu Yu-jie and Mei An (Northeast Forestry University, Harbin/China) for valuable discussions about TCM. Furthermore, we thank Dr T Paululat (Institute of Organic Chemistry, Universität Siegen/Germany) for measurement of 15N-NMR spectroscopic data.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website (http://www.nature.com/ja)
Supplementary information
Rights and permissions
About this article
Cite this article
Vollmar, D., Thorn, A., Schuberth, I. et al. A comprehensive view on 4-methyl-2-quinazolinamine, a new microbial alkaloid from Streptomyces of TCM plant origin. J Antibiot 62, 439–444 (2009). https://doi.org/10.1038/ja.2009.68
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2009.68
