
Abstract
A respiratory inhibitor, antimycin A (AA), induced an apoptotic-like cell death characterized by nuclear and DNA fragmentation in human leukemia HL-60 cells. This cell death was significantly restricted by a nitric oxide synthase (NOS) inhibitor, NG-monomethyl-L-arginine (L-NMMA), and a poly(ADP-ribose) polymerase (PARP) inhibitor, 5-aminoisoquinoline (AIQ). Indeed, NO production and PARP overactivation were detected in the cells treated with AA. On the one hand, L-NMMA partly eliminated NO production and on the other, AIQ and L-NMMA also restricted PARP activation. Excessive signals related to PARP overactivation induce the translocation of an apoptosis-inducing factor (AIF) from the mitochondria to the nuclei, resulting in DNA fragmentation. In AA-treated cells, the nuclear translocation of AIF occurred. This translocation was restricted by pretreatment with AIQ and L-NMMA. Although pretreatment with ascorbic acid eliminated the reactive oxygen species (ROS) generation induced by the blockade of complex III by AA, the pretreatment did not protect the cells from AA-induced cell death. Furthermore, cytochrome c release or caspase-3 activation was not observed in the cells treated with AA. These results suggest that AA-induced cell death does not depend on respiratory inhibition and the succeeding cascades, but on NO production, PARP overactivation and AIF translocation.
Similar content being viewed by others


Introduction
Antimycin A (AA) inhibits mitochondrial electron transport at complex III,1 thereby inhibiting respiration. The blockade of electron transport causes a collapse of the proton gradient across the mitochondrial inner membrane. This decreases mitochondrial membrane potential. It also induces the generation of reactive oxygen species (ROS).2 Both respiratory inhibition and ROS generation are reported to cause cell death in several mammalian cell lines, including mouse leukemia P388, mouse melanoma B16, human oral epidermoid carcinoma KB, human colon adenocarcinoma COLO201 and porcine renal proximal tubule LLC-PK1 cells.3, 4, 5, 6 Most of the cases of cell death induced by AA are classified as being a result of necrosis. However, there are few reports concerning the AA-induced apoptosis of LLC-PK1 and human leukemia HL-60 cells.4, 7 Although it was reported that AA promotes DNA fragmentation and nuclear and cellular disintegrations in HL-60 cells,4 the detailed mechanism of AA-induced apoptosis has been unclear. The proliferation of respiration-deficient rho0 cells of HL-60 is possible.8 This mutant generally lacks cytochrome b of complex III and various subunits of cytochrome c oxidase in addition to F1F0-ATPase.8 In addition, HL-60 cells are viable, independent of respiration. Therefore, it is unlikely that AA-induced apoptosis depends on respiratory inhibition caused by AA and the succeeding cascades. On the other hand, we recently found that AA-induced cell death accompanied with nuclear and DNA fragmentation and without cellular disintegration did not depend on the release of cytochrome c or the activation of caspase-3. In addition to these events, we also found that a nonspecific inhibitor of nitric oxide synthase (NOS), NG-monomethyl-L-arginine (L-NMMA), and an inhibitor of poly(ADP-ribose) polymerase (PARP) activation, 5-aminoisoquinoline (AIQ), significantly restricted AA-induced cell death.
Nitric oxide (NO) is formed from L-arginine and oxygen by NOS. Cytokines and stimuli, such as bacterial lipopolysaccharides induce NOS in macrophages, including HL-60 cells, which generally differentiate into macrophages in the presence of phorbol 12-myristate 13-acetate. There are many reports related to drug-induced NO production in HL-60 cells.9, 10 In addition, NO-induced apoptosis of HL-60 cells has also been reported.11, 12
PARP is an abundant nuclear enzyme that is activated by DNA strand breaks or kinks.13, 14, 15 Activated PARP consumes nicotinamide adenine dinucleotide (NAD+) to transfer poly(ADP-ribose) to specific acceptor proteins.16, 17 PARP activation contributes to DNA repair enzymes and then prevents chromatid exchange.16, 18, 19 However, excessive PARP activation can also promote cell death when extensive DNA damage occurs, as in inflammation and ischemia.17, 20 The phenomena in such cell death pathways include mitochondrial depolarization, mitochondrial permeability transition and the release of apoptosis-inducing factor (AIF) from the mitochondria. AIF is released from the mitochondria and moves to the nucleus and then triggers nuclear DNA fragmentation.
In this report, we focus on PARP-mediated cascades to clarify the biochemical process of AA-induced apoptotic-like cell death in HL-60 cells and also discuss the association of free radicals, including reactive oxygen and/or nitrogen species, produced by AA with apoptotic promotion.
Materials and methods
Cell culture
The HL-60 cell line was earlier obtained from the American Type Culture Collection (Manassas, VA, USA).21 HL-60 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum along with L-glutamine, penicillin and streptomycin at 37 °C in humidified air containing 5% CO2. The cells were subcultured twice a week, and only those in the exponential growth phase were used in the following experiments.
Antibodies and chemicals
The antibodies used were obtained from commercial sources as follows: anti-cytochrome c (BD Pharmingen, Franklin Lakes, NJ, USA); anti-actin (Sigma, St Louis, MO, USA); anti-AIF (H-300) and anti-PARP-1 (F-2) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-mouse IgG (Promega Corporation, Madison, WI, USA); and anti-rabbit IgG (Bio-Rad, Hercules, CA, USA). AA, puromycin, eukaryotic protease inhibitor cocktail, A23187, thapsigargin, Fluo3-AM, L-NMMA and AIQ were obtained from Sigma. Ac-DEVD-MCA as a caspase-3 substrate, Boc-VLK-MCA as a calpain substrate and Z-LL-H as a calpain inhibitor were obtained from Peptide Institute Inc. (Osaka, Japan). Alamar blue dye was obtained from Diagnostic Systems Inc. (Webster, TX, USA).
Cytotoxicity
Cytotoxicity was determined using Alamar blue assay, as described earlier.22 Briefly, cells (106 cells/ml) were pretreated in quadruplicate in 96-well flat-bottom tissue-culture plates with or without an antioxidant, such as ascorbic acid, for 1 h. After pretreatment, AA was added to the culture and the cells were then incubated for 24 h. After incubation with AA, Alamar blue dye was added to the culture and the cells were further incubated for another 4–8 h. Fluorescence was detected using a Tecan GENios (Männedorf, Switzerland; excitation wavelength at 530 nm, emission wavelength at 590 nm).
Measurement of ROS generation level
Cellular ROS generation was examined by a method dependent on intracellular deacylation and oxidation of 2′,7′-dichlorodihydrofluorescein-diacetate (DCFH-DA) to the fluorescent compound 2′,7′-dichlorofluorescein (DCF).23 Briefly, exponentially growing cells (106 cells/ml) were washed twice with glucose-free modified Gey's buffer (MGB; 145 mM NaCl, 5 mM KCl, 1 mM Na2HPO4, 10 mM HEPES, 1 mM CaCl2, 1 mM MgSO4, pH 7.4) and then stained in MGB with 20 μM DCFH-DA. The stained cells were incubated for 1 h. After incubation, the cells were washed with MGB. The washed cells were treated with AA or H2O2 for 1 h. Fluorescence derived from DCF was detected using a Tecan GENios (excitation wavelength at 488 nm, emission wavelength at 525 nm).
Fluorescence staining of the nuclei
Cells (106 cells/ml) were cultured with or without drugs for 24 h. The cells were washed with phosphate-buffered saline (PBS). The washed cells were fixed with 1% glutaraldehyde in PBS for 1 h and then washed with PBS twice. The fixed cells were stained with 1 mM Hoechst 33258 in PBS for 5 min. The stained cells were observed under an Olympus fluorescence microscope (Hamburg, Germany)(excitation wavelength at 365 nm, emission wavelength at 420 nm).
Analysis of oligonucleosomal DNA fragmentation
Oligonucleosomal DNA fragmentation was analyzed using a slightly modified agarose gel electrophoresis method, as described earlier.21 Cells (106 cells/ml) were cultured with or without drugs for 24 h. The cells were digested with 50 mM Tris-HCl buffer (pH 7.8) containing 10 mM EDTA, 0.5% SDS and 100 μg ml−1 proteinase K at 37 °C for 12 h. DNA was extracted with chloroform/phenol/isoamylalcohol (25:24:1, v/v), precipitated with 0.5 M NaCl/ethanol (1:1, v/v) and then electrophoresed on a 2% agarose gel. DNA fragments were stained with ethidium bromide.
Preparation of cytosolic fraction for detection of cytochrome c and AIF release
Cytochrome c and AIF release from the mitochondria was detected by western blot analysis using LAS1000 (Fujifilm, Tokyo, Japan). Cells (106 cells/ml) were cultured with or without AA in six-well flat-bottom tissue culture plates for 24 h. After cultivation, the cells were washed with a buffer containing 30 mM Tris, 75 mM sucrose, 20 mM glucose, 5 mM KH2PO4, 40 mM KCl, 0.5 mM EDTA and 3 mM MgCl2. The cells were lysed by adding 10 μM digitonin for 10 min on ice. The cell lysates were incubated with a eukaryotic protease inhibitor cocktail at 1:1000 volume of the lysates at 37 °C for 7 min. After incubation, the supernatant was harvested by centrifugation at 350 g for 3 min at 4 °C. SDS-polyacrylamide gel electrophoresis (PAGE) and western blot analysis were performed using standard protocols.
Caspase-3 and calpain assays
Caspase-3 and calpain activities were measured using fluorometric assays in which the fluorogenic synthetic peptides, Ac-DEVD-MCA and Boc-VLK-MCA, were used as substrates, respectively. Cells (106 cells/ml) were cultured with or without drugs in 48-well flat-bottom tissue culture plates for 24 h. After the cultivation, the collected cells were washed with PBS. The pellets were re-suspended in 20 mM HEPES-KOH buffer (pH 6.8) with 250 mM sucrose, 50 mM KCl, 2.5 mM MgCl2 and 1 mM dithiothreitol on ice for 10 min. The cells were lysed by adding 10 μM digitonin for 10 min on ice. The cell lysates were incubated with 14 μM Ac-DEVD-MCA in 100 mM Tris-HCl buffer (pH 7.5) or Boc-VLK-MCA in the same buffer containing 2 mM CaCl2 and 5 mM β-mercaptoethanol at 37 °C for 1 h. Fluorescence intensity was measured using a Tecan GENios (excitation wavelength at 380 nm, emission wavelength at 460 nm). One unit of enzyme activity was defined as the cleavage of 1 pmol of each substrate per minute.
Elevation of intracellular Ca2+ level
The rate of Ca2+ release was estimated by flow cytometry in which the fluorogenic dye, fluo-3 AM, was used. Cells (106 cells/ml) were cultured with 1 mg ml−1 fluo-3 AM in 48-well flat-bottom tissue culture plates for 30 min. After incubation, the collected cells were suspended in RPMI 1640 medium containing 6 mM EGTA (ethylene glycol bis(β-aminoethylether)-N,N,N′,N′,-tetraacetic acid) and 50 μM β-mercaptoethanol. The fluorescence intensity of each cell was measured using FACS Calibur, LSR (BD, Franklin Lakes, NJ, USA). Measurement of intracellular Ca2+ level was started after adding 500 μl of 6 mM EGTA. After a 30-s flow, the sample was added to 20 μl of 5 μM A23187, and the measurement was restarted.
Measurement of NO production level
Intracellular NO production level was determined by fluorometric assay using a NO assay kit (Dojindo Laboratories, Kumamoto, Japan). The principle of this kit is described as follows: NO is oxidized to NO2− and NO3− in water. The reaction of 2,3-diaminonapthalene with NO2− forms the fluorescent product, 1-(H)-napthotriazole. The fluorescence derived from 1-(H)-napthotriazole is measured (excitation wavelength at 360 nm, emission wavelength at 460 nm). In this kit, NO3− is converted into NO2− by reductase. Therefore, it is possible to estimate the total NO level using this kit. Cells (106 cells/ml) were incubated with or without drugs in 96-well flat-bottom tissue culture plates for 24 h, and 80 μl of the culture was then used for the assay.
Preparation of nuclear fraction for detection of AIF and PARP
Cells (106 cells/ml) were washed in cold PBS and suspended in 400 μl of ice-cold hypotonic buffer (10 mM HEPES/KOH, pH 7.9, 2 mM MgCl2, 0.1 mM EDTA, 10 mM KCl, 1 mM dithiothreitol, 0.5 mM PMSF (phenylmethylsulfonyl fluoride) and 1% (v/v) eukaryotic protease inhibitor cocktail) for 10 min on ice. The suspension was vortexed and centrifuged at 15 000 g for 30 s at 4 °C. The supernatant was discarded and the cell pellet was gently re-suspended in 100 μl of ice-cold saline buffer (50 mM HEPES/KOH (pH 7.9), 50 mM KCl, 300 mM NaCl, 0.1 mM EDTA, 10% glycerol, 1 mM DTT, 0.5 mM PMSF, 1% (v/v) eukaryotic protease inhibitor cocktail) on ice for 20 min. The cell suspension was vortexed and centrifuged at 15 000 g for 5 min at 4 °C. The supernatant was stored at −70 °C as a nuclear lysate.
Determination of intracellular NAD+ level
The NAD+ concentration was determined as described with a slight modification.24 Briefly, 106 cells were re-suspended in 100 μl of 0.5 M perchloric acid. The obtained cell extracts were neutralized with equal volumes of 1 M KOH and 0.33 M KH2PO4/K2HPO4 (pH 7.5). After centrifugation, to remove the KClO4 precipitate, 200 μl of NAD+ reaction mixture (600 mM ethanol, 0.5 mM 3-[4,5 dimethylthiazol-2-yl]-2,5 diphenyltetrazolium bromide, 2 mM phenazine ethosulfate, 5 mM EDTA, 1 mg ml−1 bovine serum albumin, 120 mM bicine at pH 7.8) was added to 50 μl of the supernatant or NAD+ standard, and the mixture was incubated for 5 min at 37 °C. Alcohol dehydrogenase (25 μl) was added to the reaction mixture and the resulting mixture was incubated for 20 min at 37 °C. To stop the reaction, 250 μl of 12 mM iodoacetate was added, and the optical density was then read at 570 nm.
Measurement of intracellular ATP level
Intracellular ATP level was determined by luminescence assay using D-luciferin and luciferase (Toyo Ink, Tokyo, Japan). Cells (106 cells/ml) were incubated with or without drugs for 24 h in 96-well flat-bottom tissue culture plates. After incubation, the washed cells were suspended in PBS and 100 μl of luminous dye was then added. After 10 min, the luminescence was detected using a Tecan GENios.
Analysis of membrane potential
The fluorescent stain rhodamine 123 was used as a probe to assess the mitochondrial membrane potential of living HL-60 cells. Membrane potential analysis was described earlier.25 Cells were treated with 50 μg ml−1 AA or 25 μM carbonyl cyanide m-chlorophenylhydrazone (CCCP) for 24 h.
Statistical analysis
Values represent means±s.d. of at least three independent experiments, unless otherwise indicated.
Results
Cytotoxicity of AA and AA-induced ROS generation in HL-60 cells
After exposure to AA for 24 h, the cells showed a dose-dependent cytotoxicity against AA at concentrations higher than 3 μg ml−1 (Figure 1a). In addition, at the concentrations of 0.2–3 μg ml−1, AA also showed a weak cytotoxicity. The IC50 of AA was 10 μg ml−1. This IC50 was considered to be higher than those of AA in other cell lines, such as P388 B16, and KB.3 ROS generation was evaluated as DCFH-DA oxidation. This probe was highly reactive with hydrogen peroxide and has been used in evaluating ROS generation in mammalian cells.23 AA and all the antioxidants tested did not directly react with DCFH-DA (data not shown). In addition, pretreatment of the cells with all the antioxidants tested did not result in ROS generation (Figure 1b). The exposure of HL-60 cells to AA for 1 h induced ROS generation mainly as hydrogen peroxide (Figure 1b). Antioxidants restrict the oxidative damage to cellular components caused by ROS. Indeed, pretreatment with a hydrophilic ascorbic acid prevented nearly all the AA-induced ROS generation (Figure 1b). Other antioxidants, such as α-tocopherol and N-acetyl cysteine, showed no effect. On the other hand, pretreatment with ascorbic acid hardly altered the cytotoxicity of AA (Figure 1c).

Effects of AA on HL-60 cells. (a) Cytotoxicity of AA. After the treatment of HL-60 cells with AA for 24 h, cytotoxicity was evaluated using Alamar blue assay. (b) ROS generation induced by AA. HL-60 cells were pretreated with or without 25 μM of each antioxidant for 1 h. Addition of no drug (None), 50 μg ml−1 AA (AA) or 10 μg ml−1 hydrogen peroxide (H2O2) into the culture was carried out, and the cells were further incubated for 1 h before the measurement of ROS generation level. ROS generation was defined as 100% in the control experiment, in which incubation was carried out without antioxidant and AA. (c) Cytotoxicity of AA under a suppressed condition of ROS. HL-60 cells were pretreated with 0 or 25 μM ascorbic acid for 1 h. After pretreatment, AA was added to the culture and the cells were then incubated for 24 h. Cytotoxicity was evaluated using Alamar blue assay. (d) Oligonucleosomal DNA fragmentation induced by AA. HL-60 cells were treated with 50 μg ml−1 AA or 10 μg ml−1 puromycin for 24 h. (e) Nuclear fragmentation induced by AA. HL-60 cells were treated for 24 h with 50 μg ml−1 AA or 10 μg ml−1 puromycin. The fluorescence staining of nuclei was performed using 200 μM Hoechst 33258 for 5 min.
AA-induced apoptotic-like cell death of HL-60 cells
In AA-treated cells, nuclear fragmentation, chromatin condensation and formation of apoptotic bodies were observed, as well as in those treated with a typical apoptotic inducer, puromycin, which inhibits cytoplasmic protein synthesis (Figure 1e). Although the puromycin-treated cells showed cellular disintegration, most of the AA-treated cells did not show such effects. Oligonucleosomal DNA fragments were also detected in the DNA fraction extracted from AA-treated cells (Figure 1d).
Cytochrome c release from the mitochondria and caspase-3 activity
AA did not activate caspase-3 (Figure 2a). Interestingly, the activity of caspase-3 was significantly lower than that in the case of the drug-free control. On the other hand, puromycin activated caspase-3.

Effects of AA on mitochondrial- and ER-related apoptotic responses of HL-60 cells. (a) No activation of caspase-3 in HL-60 cells was treated with AA. HL-60 cells treated with 50 μg ml−1 AA or 10 μg ml−1 puromycin for 24 h. None indicates no treatment with drugs. (b) No release of cytochrome c from the mitochondria in HL-60 cells treated with AA. HL-60 cells were treated with no drug, 50 μg ml−1 AA or 25 μM carbonyl cyanide m-chlorophenylhydrazone (CCCP) for 24 h followed by western blot analysis as described in the Materials and Methods section. β-Actin was used as an internal control to show that the amounts of proteins subjected to SDS-PAGE were the same. Data are representative of one of three independent experiments. (c) Slight increase in intracellular Ca2+ level induced by AA. HL-60 cells were incubated with AA at 0 (None) or 50 (AA) μg ml−1 for 24 h. The washed cells were stained with 5 μg ml−1 Fluo3-AM and then analyzed by flow cytometry to estimate the intracellular Ca2+ level. (d) Calpain activation induced by AA. HL-60 cells were pretreated with 0 (None, AA and TG) or 25 (AA+Z-LL-H) μM, calpain inhibitor, Z-LL-H, for 1 h. After pretreatment, no drug (None), 50 μg ml−1 AA (AA and AA+Z-LL-H) or 2 μg ml−1 thapsigargin (TG) was added to the culture and the cells were then incubated for 24 h. (e) No restoration of AA-induced cytotoxicity by the calpain inhibitor. HL-60 cells were pretreated with 0 (AA) or 25 (AA+Z-LL-H) μM Z-LL-H for 1 h. After pretreatment, AA was added to the culture and the cells were then incubated for 24 h before Alamar blue assay.
CCCP, which induces the depolarization of mitochondrial inner membrane, apparently caused the release of cytochrome c. In the case of AA treatment, the level of cytochrome c release from the mitochondria was similar to that in the control (Figure 2b).
Intracellular Ca2+ level and calpain activity
In a positive control, a calcium ionophore, A23187, induced a massive increase in intracellular Ca2+ level immediately after its addition (data not shown). On the other hand, AA slightly increased intracellular Ca2+ level after 24-h incubation (Figure 2c). This observation indicates that AA is not merely a calcium ionophore rather it promotes a slight Ca2+ release by a mechanism different from that of A23187.
The activation of calpains is also stimulated by endoplasmic reticulum (ER) stress by Ca2+ release into the cytosol.7 Thapsigargin, which is an ER Ca2+-ATPase inhibitor, increased calpain activity (Figure 2d). In addition, AA significantly increased calpain activity. Furthermore, pretreatment with the calpain inhibitor, Z-LL-H, significantly suppressed the AA-induced increase in calpain activity (Figure 2d). However, pretreatment with the inhibitor did not decrease the cytotoxicity of AA (Figure 2e).
NO production
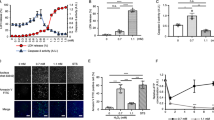
In the pretreatment with L-NMMA, which is a nonspecific inhibitor of NOS, a four-fold increase in the IC50 of AA was observed, indicating that the cytotoxicity of AA was significantly restricted by pretreatment with L-NMMA (Figure 3a). Moreover, we detected NO production in AA-treated cells (Figure 3b). AA did not directly react with 2,3-diaminonapthalene as a probe for NO detection (data not shown). In addition, pretreatment with L-NMMA decreased the NO production level of AA-treated cells by 55%. On the other hand, a slight decrease in NO production level was observed in AA-treated cells pretreated with N-acetyl cysteine.

Effects of AA on NO production and PARP-related apoptotic responses of HL-60 cells. (a) Restoration of AA-induced cytotoxicity by a NOS inhibitor L-NMMA. HL-60 cells were pretreated with 0 (AA) or 1 (AA+L-NMMA) mM L-NMMA for 1 h. After pretreatment, AA was added to the culture and the cells were then incubated for 24 h before Alamar blue assay. (b) NO production in AA-treated cells. HL-60 cells were pretreated with or without 1 mM L-NMMA and 5 mM N-acetyl cysteine for 1 h. After pretreatment, AA at 0 (None) or 50 (AA) μg ml−1 was added to the culture and the cells were further incubated for 24 h. (c) Restoration of AA-induced cytotoxicity by the PARP inhibitor AIQ. HL-60 cells were pretreated with 0 (AA) or 10 (AA+AIQ) nM AIQ for 1 h. After pretreatment, AA was added to the culture and the cells were further incubated for 24 h before Alamar blue assay. (d) Detection of AIF in the cytoplasm and the nuclei, and activation of PARP-1 induced by AA. HL-60 cells were pretreated with no inhibitor (N and A), 10 nM AIQ (I) and 1 mM L-NMMA (L) for 1 h. After pretreatment, 0 (N) or 50 (A, I and L) μg ml−1 AA was added to the culture and the cells were then incubated for 24 h followed by western blot analysis as described in the Materials and methods section. β-Actin was used as an internal control to show that the amounts of proteins subjected to SDS-PAGE were the same. Data are representative of one of three independent experiments.
Detection of AIF release and cleaved PARP-1
Pretreatment with AIQ, which restricts the activation of PARP,26 significantly restricted the cytotoxicity of AA (Figure 3c), indicating that the progression of AA-induced cell death depends on PARP activation. AIF directly translocates from the mitochondrial inner membrane to the nucleus and then participates in the execution of DNA fragmentation. This is stimulated by activated calpains or PARP cycle.27, 28
The release of AIF into the cytosol and its translocation into the nuclei were not detected in a control experiment (Figure 3d). On the other hand, AA induced the migration of AIF to the nuclei (Figure 3d). In addition, in the case of pretreatment with AIQ, 45 and 30% decreases in cytosolic and nuclear AIF levels, respectively, were observed after AA treatment. Moreover, in the case of pretreatment with L-NMMA, 70 and 63% decreases in cytosolic and nuclear AIF levels, respectively, were observed after AA treatment. These indicate that the AA-induced AIF migration was partly suppressed in the cells pretreated with AIQ and L-NMMA. The cleaved PARP-1, which is an activated form, was detected in cells treated with AA. In addition, this activation was clearly inhibited by pretreatment with AIQ and L-NMMA (Figure 3d).
Intracellular NAD+ and ATP levels
Decreases in the NAD+ and ATP levels were observed in AA-treated cells (Figures 4a and b). PARP activation results in a rapid decline in the levels of cellular NAD+ and ATP as substrates.27 These results suggest that AA-induced apoptosis is involved in PARP activation. Furthermore, the NAD+ level in AA-treated cells pretreated with AIQ and L-NMMA was partly but significantly restored as compared with that in cells treated with only AA (Figure 4a). On the other hand, the ATP level in AA-treated cells pretreated with AIQ and L-NMMA was slightly restored to the control level (Figure 4b).

Effects of AA on NAD+ and ATP levels, and membrane potential on HL-60 cells. (a) Reduction of intracellular NAD+ level induced by AA. HL-60 cells were pretreated with no inhibitor (None and AA), 10 nM AIQ (AA+AIQ) and 1 mM L-NMMA (AA+L-NMMA) for 1 h. After pretreatment, AA at 0 (None) or 50 (AA, AA+AIQ and AA+L-NMMA) μg ml−1 was added to the culture and the cells were then incubated for 24 h. (b) Decrease in intracellular ATP level induced by AA. HL-60 cells were pretreated with 0 (None and AA), 10 nM AIQ (AA+AIQ) and 1 mM L-NMMA (AA+L-NMMA) for 1 h. After pretreatment, AA at 0 (None) or 50 (AA, AA+AIQ and AA+L-NMMA) μg ml−1 was added to the culture and the cells were then incubated for 24 h. (c) Loss of the mitochondrial membrane potential induced by AA. HL-60 cells were pretreated with 0 (None, AA and CCCP), 10 nM AIQ (AA+AIQ) and 1 mM L-NMMA (AA+ L-NMMA) for 1 h. After pretreatment, AA at 0 (None) or 50 (AA, AA+AIQ and AA+L-NMMA) μg ml−1, or 10 μM CCCP (CCCP) was added to the culture and the cells were then incubated for 24 h.
Mitochondrial membrane potential of AA-treated cells
The release of AIF from the mitochondria is caused by the decrease in mitochondrial membrane potential.29 We detected the decrease in the mitochondrial membrane potential in AA-treated cells. The decrease in the membrane potential of AA-treated cells was similar to that of CCCP-treated cells (Figure 4c). In addition, L-NMMA and AIQ significantly restricted the AA-induced decrease in the membrane potential.
Discussion
AA specifically binds to the bc1 complex in the mitochondrial electron transport chain, thereby inhibiting respiration and then inducing ROS generation. Cell death induced by AA was classified as mostly being a result of necrosis and partially being a result of apoptosis. In many cell lines, the toxic effect of AA is extremely strong; the range of IC50 was 0.01–0.2 μg ml−1.30 This is probably directly caused by the inhibition of respiration and ROS generation at complex III. On the other hand, AA dose dependently induced the death of HL-60 cells at concentrations higher than 3 μg ml−1. The IC50 was 10 μg ml−1 (Figure 1a). It is possible to obtain respiration-deficient rho0 cells of HL-60,8 indicating proliferation without oxygen respiration. Although AA-induced ROS generation was eliminated in HL-60 cells by pretreatment with ascorbic acid (Figure 1b), the pretreatment hardly altered the toxicity of AA (Figure 1c). Therefore, for AA-induced cell death, factors other than respiratory inhibition or its indirect or secondary effects, such as ROS generation, are considered in the case of HL-60 cells. Nuclear and DNA fragmentations were detected, but cellular disintegration was not observed in the AA-treated cells, indicating that AA induced a type of apoptotic-like cell death.
Many stimuli are transmitted as a cascade of apoptotic execution signals starting from the mitochondria, namely the signals generated by various environmental changes are consolidated into the mitochondria first, then cytochrome c and AIF are released from the mitochondria.31 Interestingly, AA did not cause the release of cytochrome c but that of AIF in HL-60 cells (Figures 2b and 3c). This is supported by a report that AA prevents NO-induced apoptosis by blocking cytochrome c release independent of Bcl-2 expression in the rat gastric mucosa cell line RGM-1.32 This apoptosis was triggered by a pure NO donor, 1-hydroxy-2-oxo-3,3-bis(2-aminoethyl)-1-triazene. On the other hand, we found that treatment with a high concentration of AA, such as 50 μg ml−1, induced cellular NO production in HL-60 cells (Figure 3b).
AIF translocation from the mitochondria to the nuclei occurs through PARP activation when Ca2+ influx was observed. Ca2+ influx activates calcium-dependent cytoplasmic proteins, such as neuronal NOS, in the brain. NO produced in the presence of increased levels of ROS leads to peroxynitrite formation, resulting in oxidative DNA damage. Excessive DNA damage overactivates PARP. PARP activation and/or its related signals stimulate the translocation of AIF from the mitochondria to the nuclei through the loss of mitochondrial membrane potential, resulting in nuclear apoptosis. The loss of mitochondrial membrane potential is reported to trigger the release of AIF.33 We showed the loss of mitochondrial membrane potential in AA-treated cells (Figure 4c).
L-NMMA is an inhibitor of three isotypes of NOS (endothelial NOS, neuronal NOS and inducible NOS). This inhibitor partly eliminated NO production but significantly restricted the cytotoxicity of AA (Figure 3a). The protective effect of this inhibitor suggests that NO induced the death of AA-treated HL-60 cells. In HL-60 cells, the activation of inducible NOS was reported.12 Therefore, it may be considered that AA-induced cell death is involved in NO production by inducible NOS.
Caspase-3 is activated in apoptotic inductions dependent on cytochrome c release. The AA-induced cell death did not depend on a cascade by the release of cytochrome c from the mitochondria or the activation of caspase-3 (Figures 2a and b), indicating the possibility that this apoptotic execution occurs only through AIF release.
In nuclear extracts of AA-treated cells, we detected cleaved PARP-1, or its active form, and AIF (Figure 3c). In addition, AIQ, which is an inhibitor of PARP activation, restricted the cytotoxicity of AA, PARP activation and AIF translocation. In addition, in AA-treated cells, both NAD+ and ATP levels were lower than those in control cells. The NAD+ level was partly restored by AIQ and L-NMMA (Figure 4a). These results support the observed AA-induced PARP-1 activation. On the other hand, the ATP level was slightly restored by AIQ and L-NMMA (Figure 4b). This result indicates that the cause of the decreased ATP level in AA-treated cells might be factors such as the inhibition of the electron transport chain and/or unknown mechanisms.
ER stress-induced apoptosis could be induced by only AIF release without cytochrome c release from the mitochondria.34 When ER experiences potent stress, Ca2+ is released into the cytosol from ER, and calpains are then activated. Although the intracellular Ca2+ level was slightly elevated (Figure 2c) and calpains were strongly activated in the cells treated with AA (Figure 2d), the calpain inhibitor used hardly decreased the cytotoxicity of AA (Figure 2e). These results suggest that the AA-induced cell death is not due to ER stress.
Recently, another target of AA has been reported. AA interacts with the BH3 domain (Bcl-2 homology domain 3)-binding hydrophobic groove of anti-apoptotic proteins Bcl-2 and Bcl-xL.35 The BH3 domain is present in the Bcl proteins that promote and restrict apoptotic cell death. AA induces the loss of mitochondrial membrane potential in addition to the mitochondria overexpressing Bcl-2 and Bcl-xL, thereby probably inducing the apoptosis of the murine hepatocyte cell line TAMH. Bcl-2 and Bcl-xL restrict cytochrome c release.36 This result is in conflict with our results, wherein AA did not cause the release of cytochrome c. On the other hand, there is a report that AA prevents cytochrome c release independent of Bcl-2 expression when NO-induced apoptosis is suppressed by AA.32 This is consistent with our results.
In conclusion, our results taken together showed that AA induced cellular NO production, PARP overactivation and translocation of AIF from the mitochondria to the nuclei, resulting in the apoptotic-like cell death of HL-60. Further investigations are required to clarify the mechanism by which cytochrome c release from the mitochondria is restricted by AA.
References
Pham, N. A., Robinson, B. H. & Hedley, D. W. Simultaneous detection of mitochondrial respiratory chain activity and reactive oxygen in digitonin-permeabilized cells using flow cytometry. Cytometry 41, 245–251 (2004).
Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J. Biol. Chem. 267, 8834–8839 (1992).
Kaushal, G. P., Ueda, N. & Shah, S. V. Role of caspases (ICE/CED 3 proteases) in DNA damage and cell death in response to a mitochondrial inhibitor, antimycin A. Kidney Int. 52, 438–445 (1997).
Malcolm, A. K. & Monica, A. Antimycin A-induced apoptosis of HL-60 cells. Cytometry 49, 106–112 (2002).
Takimoto, H., Machida, K., Ueki, M., Tanaka, T. & Taniguchi, M. UK-2A, B, C and D, novel antifungal antibiotics from Streptomyces sp. 517-02. IV. Comparative studies of UK-2A with antimycin A3 on cytotoxic activity and reactive oxygen species generation in LLC-PK1 cells. J. Antibiot. 52, 480–484 (1999).
Fujita, K., Kiso, T., Usuki, Y., Tanaka, T. & Taniguchi, M. UK-2A, B, C and D, novel antifungal antibiotics from Streptomyces sp. 517-02 VI (3). Role of substituents on dilactone ring of UK-2A and antimycin A3 against generation of reactive oxygen species in porcine renal proximal tubule LLC-PK1 cells. J. Antibiot. 57, 687–690 (2004).
Broker, L. E., Kruyt, F. A. & Giaccone, G. Cell death independent of caspases: a review. Clin. Cancer Res. 11, 3155–3162 (2005).
Hail, N. Jr. & Lotan, R. Apoptosis induction by the natural product cancer chemopreventive agent deguelin is mediated through the inhibition of mitochondrial bioenergetics. Apoptosis 9, 437–447 (2004).
Everett, S. A. et al. Nitric oxide involvement in the toxicity of hydroxyguanidine in leukaemia HL60 cells. Br. J. Cancer Suppl. 27, S172–S176 (1996).
Oguro, A., Kawase, T. & Orikasa, M. NaF induces early differentiation of murine bone marrow cells along the granulocytic pathway but not the monocytic or preosteoclastic pathway in vitro. In Vitro Cell Dev. Biol. Anim. 39, 243–248 (2003).
Laouar, A., Glesne, D. & Huberman, E. Protein kinase C-beta, fibronectin, alpha(5)beta(1)-integrin, and tumor necrosis factor-alpha are required for phorbol diester-induced apoptosis in human myeloid leukemia cells. Mol. Carcinog. 32, 195–205 (2001).
Bhushan, S. et al. A triterpenediol from Boswellia serrata induces apoptosis through both the intrinsic and extrinsic apoptotic pathways in human leukemia HL-60 cells. Apoptosis 12, 1911–1926 (2007).
de Murcia, G. et al. Structure and function of poly(ADP-ribose) polymerase. Mol. Cell. Biochem. 138, 15–24 (1994).
de Murcia, G. & Ménissier-de Murcia, J. Poly(ADP-ribose) polymerase: a molecular nick-sensor. Trends Biochem. Sci. 19, 172–176 (1994).
Lautier, D., Lagueux, J., Thibodeau, J., Ménard, L. & Poirier, G. G. Molecular and biochemical features of poly(ADP-ribose) metabolism. Mol. Cell. Biochem. 122, 171–193 (1993).
D'Amours, D., Desnoyers, S., D'Silva, I. & Poirier, G. G Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 342, 249–268 (1999).
Virág, L. & Szabó, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 54, 375–429 (2002).
Bryant, H. E. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005).
Oei, S. L., Keil, C. & Ziegler, M. Poly(ADP-ribosylation) and genomic stability. Biochem. Cell Biol. 83, 263–269 (2005).
Eliasson, M. J. et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 3, 1089–1095 (1997).
Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 15, 2922–2933 (2001).
Kiso, T., Usuki, Y., Ping, X., Fujita, K. & Taniguchi, M. L-2,5-dihydrophenylalanine, an inducer of cathepsin-dependent apoptosis in human promyelocytic leukemia cells (HL-60). J. Antibiot. 54, 810–817 (2001).
Carter, W. O., Narayanan, P. K. & Robinson, J. P. Intracellular hydrogen peroxide and superoxide anion detection in endothelial cells. J. Leukoc. Biol. 55, 253–258 (1994).
Zong, W. X., Ditsworth, D., Bauer, D. E., Wang, Z. Q. & Thompson, C. B. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 18, 1272–1282 (2004).
Burger, A. M. et al. Tyrphostin AG17, [(3,5-Di-tert-butyl-4-hydroxybenzylidene)-malononitrile], inhibits cell growth by disrupting mitochondria. Cancer Res. 55, 2794–2799 (1995).
Szabó, G. et al. Poly(ADP-ribose) polymerase inhibition reduces reperfusion injury after heart transplantation. Circ. Res. 90, 100–106 (2002).
Yu, S. W., Wang, H., Dawson, T. M. & Dawson, V. L. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol. Dis. 14, 303–317 (2003).
Polster, B. M., Basanez, G., Etxebarria, A., Hardwick, J. M. & Nicholls, D. G. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J. Biol. Chem. 280, 6447–6454 (2005).
Hong, S. J., Dawson, T. M. & Dawson, V. L. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol. Sci. 25, 259–264 (2004).
Ueki, M. et al. UK-3A, a novel antifungal antibiotic from Streptomyces sp. 517-02: fermentation, isolation, structural elucidation and biological properties. J. Antibiot. 50, 551–555 (1997).
Ferri, K. F. & Kroemer, G. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 3, E255–E263 (2001).
Dairaku, N. et al. Oligomycin and antimycin A prevent nitric oxide-induced apoptosis by blocking cytochrome c leakage. J. Lab. Clin. Med. 143, 143–151 (2004).
Susin, S. A. et al. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 192, 571–580 (2000).
Newcomb, E. W., Lukyanov, Y., Smirnova, I., Schnee, T. & Zagzag, D. Noscapine induces apoptosis in human glioma cells by an apoptosis-inducing factor-dependent pathway. Anticancer Drugs 19, 553–563 (2008).
Tzung, S. P. et al. Antimycin A mimics a cell-death-inducing Bcl-2 homology domain 3. Nat. Cell Biol. 3, E43–E46 (2001).
Yang, J. et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275, 1129–1132 (1997).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ogita, M., Ogita, A., Usuki, Y. et al. Antimycin A-induced cell death depends on AIF translocation through NO production and PARP activation and is not involved in ROS generation, cytochrome c release and caspase-3 activation in HL-60 cells. J Antibiot 62, 145–152 (2009). https://doi.org/10.1038/ja.2009.2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2009.2
Keywords
This article is cited by
-
Oleoylethanolamide protects mesenchymal stem/stromal cells (MSCs) from oxidative stress and reduces adipogenic related genes expression in adipose-derived MSCs undergoing adipocyte differentiation
Molecular Biology Reports (2024)
-
Antimycin A induced apoptosis in HCT-116 colorectal cancer cells through the up- and downregulation of multiple signaling pathways
Medical Oncology (2022)
