
Abstract
Chlamydiae are obligate intracellular bacteria comprising important human pathogens and symbionts of protists. Molecular evidence indicates a tremendous diversity of chlamydiae particularly in marine environments, yet our current knowledge is based mainly on terrestrial representatives. Here we provide first insights into the biology of marine chlamydiae representing three divergent clades. Our analysis of single-cell amplified genomes revealed hallmarks of the chlamydial lifestyle, supporting the ancient origin of their characteristic developmental cycle and major virulence mechanisms. Surprisingly, these chlamydial genomes encode a complete flagellar apparatus, a previously unreported feature. We show that flagella are an ancient trait that was subject to differential gene loss among extant chlamydiae. Together with a chemotaxis system, these marine chlamydiae are likely motile, with flagella potentially playing a role during host cell infection. This study broadens our view on chlamydial biology and indicates a largely underestimated potential to adapt to different hosts and environments.
Similar content being viewed by others


Introduction
Investigating environmental relatives of bacterial pathogens has been fundamental for understanding their evolution and biology, and the emergence of virulence (Horn, 2008; Gordon et al., 2009; Robins and Mekalanos, 2014; Khodr et al., 2016; Maury et al., 2016). The chlamydiae include some of the most successful bacterial pathogens of humans, such as Chlamydia trachomatis causing over 100 million infections worldwide each year (Wright et al., 2008; Darville, 2013; Roulis et al., 2013; Dal Conte et al., 2014; Knittler and Sachse, 2015). They likely represent the most ancient group of obligate intracellular bacteria and are characterized by a biphasic developmental cycle consisting of the replicative intracellular reticulate body and the infectious extracellular elementary body (Greub and Raoult, 2003; Horn et al., 2004; Schmitz-Esser et al., 2004; Abdelrahman and Belland, 2005; Collingro et al., 2011; Nunes and Gomes, 2014). Chlamydiae generally possess reduced genomes, lack essential biosynthesis pathways and thus rely on a variety of metabolites from their eukaryotic hosts (Omsland et al., 2014).
No free-living chlamydia has been identified to date, but chlamydiae are frequently found within protists and arthropods that are ubiquitous in terrestrial environments (Horn, 2008; Taylor-Brown et al., 2015). Chlamydiae have also been identified in a number of marine animals, including fish in which they have been associated with the gill disease epitheliocystis (Karlsen et al., 2008; Draghi et al., 2010; Mitchell et al., 2010; Fehr et al., 2013; Stride et al., 2013; Nylund et al., 2015; Steigen et al., 2015; Pawlikowska-Warych and Deptuła, 2016). Moreover, metagenomics and amplicon sequencing data indicates the existence of a vast, yet unexplored diversity of divergent chlamydiae, primarily in marine environments (Pizzetti et al., 2012; Lagkouvardos et al., 2014; Pizzetti et al., 2015; Vanthournout and Hendrickx, 2015; Bou Khalil et al., 2016). However, owing to their obligate intracellular lifestyle our knowledge about chlamydiae is based on a limited number of representatives, originating exclusively from non-marine environments.
Single-cell sorting in combination with genome amplification and sequencing has been used successfully to explore yet uncultured microbes (Stepanauskas, 2012; Rinke et al., 2013). Here, we applied this approach to study the biology of marine chlamydiae. Our findings suggest that while basic features of the chlamydial lifestyle are well conserved, marine chlamydiae show unique adaptations, indicating that the biology of chlamydiae is much more variable than currently recognized.
Materials and methods
Sampling and sequencing of single-cell amplified genomes
Samples were collected from 100, 150 and 185 m depth intervals in Saanich Inlet a seasonally anoxic fjord on Vancouver Island, British Columbia (Station S3 48°35′18.0″N, 123°30′13.2″W), on 9 August 2011. Sample collection and biochemical measurements were performed as previously described (Zaikova et al., 2010; Roux et al., 2014). Water column redox conditions were typical for stratified summer months. One milliliter aliquots were amended with 5% glycerol and 1 × TE buffer (all final concentrations), and stored at −80 °C until further processing. Sediment samples were collected in October 2014 from the upper 5 cm of sublittoral sediment off the island of Sylt (North Sea, Germany, 55°05′38.9″N, 8°15′48.8″E). After immediate transfer to the lab, the sediment was mixed and 1 ml was transferred to a 50 ml plastic tube. After adding 3 ml of sterile-filtered seawater, slurries were vortexed at maximum speed for 3 min. Sand grains were allowed to settle and the supernatant was filtered through a 5 μm pore-size membrane. The cell extracts were cryopreserved with N,N,N-trimethylglycine (‘glycine betaine’) (Sigma-Aldrich, St Louis, MO, USA) at a final concentration of 4% according to Cleland et al.(2004), and stored at −80 °C until further processing.
Single-cell sorting and whole-genome multiple displacement amplification were performed at the Bigelow Laboratory Single Cell Genomics Center (https://scgc.bigelow.org; SCGC) as described previously (Swan et al., 2011). For Saanich Inlet, a total of 315 single cells per sample were subjected to multiple displacement amplification and the taxonomic identity of single amplified genomes (SAGs) was determined by directly sequencing 16S rRNA gene amplicons at the SCGC. A single SAG (SAG AB-751-O23) from 150 m was affiliated with the phylum Chlamydiae. A 500±50 bp shotgun library was created with Nextera XT (Illumina) reagents and sequenced with NextSeq 500 (Illumina) in 2 × 150 bp mode using v.1 reagents. The obtained sequence reads were quality-trimmed with Trimmomatic v0.32 4 using the following settings: -phred33 LEADING:0 TRAILING:5 SLIDINGWINDOW:4:15 MINLEN:36. Human DNA (⩾95% identity to the Homo sapiens reference assembly GRCh38) and low complexity reads (containing <5% of any nucleotide) were removed. The remaining reads were digitally normalized with kmernorm 1.05 (http://sourceforge.net/projects/kmernorm) and then assembled with SPAdes v.3.0.0. Each end of the obtained contigs was trimmed by 100 bp, and then only contigs longer than 2000 bp were retained. The accuracy of the resulting assemblies was assessed by applying the same workflow on previously cultured and sequenced strains of Prochlorococcus marinus and Escherichia coli and comparing the obtained benchmark SAG assemblies against reference genomes with QUAST (Gurevich et al., 2013), see public benchmark data sets on the SCGC website. North Sea SAGs were screened for 16S rRNA genes by PCR using primers 341 f and 907rev (Muyzer et al., 1993; Muyzer and Smalla, 1998). For the two SAGs affiliated with chlamydiae (AG-110-P3 and AG-110-M15) 500 bp shotgun libraries were created with the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB, Frankfurt, Germany). These libraries were sequenced with a HiSeq2500 (Illumina) in 2 × 250 bp rapid mode at the Max Planck Genome Centre (MP-GC; Cologne, Cologne, Germany, http://mpgc.mpipz.mpg.de/home/). Reads were trimmed and adaptors were removed with bbduk (BBMap-35.07 suite, https://sourceforge.net/projects/bbmap) using a minimum quality cutoff of 25 and checked for quality with FastQC (Version 0.11.3, http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). Reads were assembled de novo using SPAdes 3.6.2 (Bankevich et al., 2012) in single-cell mode (k-mer sizes: 21, 35, 43, 55, 63, 71, 81, 91, 99). Then all reads were remapped against the genome scaffold (minimum sequence identity of 97%) and reassembled iteratively with SPAdes 3.6.2. (Bankevich et al., 2012) as described previously (Mußmann et al., 2017). Contigs larger than 2000 bp were retained and assembly quality was assessed with QUAST 3.0 (Gurevich et al., 2013).
Genome annotation and analysis
For genome analysis only contigs >2 kb were used unless explicitly mentioned. The quality and contamination of all three assembled SAGs was controlled with CheckM 1.0.6 (Parks et al., 2015). Automatic genome annotation was performed with ConsPred 1.24 (Weinmaier et al., 2016). The chlamydial origin of the SAGs was verified with CheckM 1.0.6, the taxonomic affiliation of the first blast hit of each CDS against the GenBank nr database (as in December 2015) (Camacho et al., 2009; Benson et al., 2013), and the presence of chlamydial core genes. The chlamydial core gene set was determined by OrthoMCL (version 2.0.9) clustering (Li et al., 2003) of proteins encoded in 26 genomes from members of seven chlamydial families. Taxonomic profiles and gene distributions were illustrated with Circos 0.69 (Krzywinski et al., 2009). The synteny of flagellar and chemotaxis genes were visualized with the GenoPlotR package in R (Guy et al., 2010). Affiliation of proteins encoded in the SAGs with clusters of orthologous groups was determined by blast against the eggNOG database (as in December 2015) (Huerta-Cepas et al., 2016). Prediction of potential effector proteins of the NF-T3SSs was performed using the respective tool at EffectiveDB (Eichinger et al., 2016).
Phylogenetic analyses of 16S rRNA genes, ribosomal and flagellar proteins
Full-length 16S rRNA gene sequences of chlamydiae were downloaded from GenBank and aligned to the SILVA SSU Ref database containing preconfigured high-quality, full-length sequences in ARB (Ludwig et al., 2004; Quast et al., 2013). Bayesian inference analysis of 16S rRNA gene sequences was carried out with MrBayes 3.2.6 using standard settings via the CIPRES Science Gateway (Miller et al., 2010; Ronquist et al., 2012). The partial 16S rRNA gene sequence of AG-110-M15 was added subsequently to the Bayesian tree using the Quick-Add Parsimony option in ARB (Ludwig et al., 2004).
A concatenated set of seven ribosomal proteins and single-copy marker genes present in all three SAGs (including rl6, rl18, rs5, rs7, rs10, rs12 and Ef-Tu) was used to verify the affiliation of the SAGs with the chlamydiae and to exclude potential contamination of the assemblies. The data set included sequences from available chlamydial genomes as well as other members of the PVC superphylum, which includes the most closely related free-living relatives of chlamydiae (Wagner and Horn, 2006). Single proteins were aligned with MUSCLE 3.8.31 (Edgar, 2004) prior to concatenation. A Bayesian tree was calculated with MrBayes 3.2.6 using the mixed amino acid model and standard settings via the CIPRES Science Gateway (Miller et al., 2010; Ronquist et al., 2012).
Protein sequences encoding components of the flagellar apparatus were identified with blast and extracted from genome sequence entries at GenBank or RefSeq. The data set included genomes used by Abby and Rocha (2012), available chlamydial genome sequences as well as selected genome sequences from the Planctomycetes and Verrucomicrobia comprising 126 genome sequences in total (Supplementary Table S3). In addition, FliPQR amino acid sequences from the SAG AB-751-O23 were extracted from contigs smaller than 2 kb, which were otherwise not considered in this study. All protein sequences were aligned with MUSCLE 3.8.31 (Edgar, 2004). Maximum likelihood phylogenies were calculated with RAxML 8.2.8 with 100 bootstrap iterations using PROTGAMMAAUTO for automatic protein model selection (Stamatakis, 2014) at the Life Science Compute Cluster at the University of Vienna, and Bayesian inference was performed using MrBayes 3.2.6 using the mixed amino acid model and standard settings via the CIPRES Science Gateway (Miller et al., 2010; Ronquist et al., 2012). Phylogenetic trees were visualized with iTOL (Letunic and Bork, 2016).
Presence of chlamydial FlhA proteins in metagenomic data sets
Around two million FlhA amino acid sequences from metagenomic data sets available at the IMG/M database in December 2015 (Markowitz et al., 2012) excluding metagenomes from human and mouse microbiomes were downloaded. Only sequences longer than 450 amino acids were kept and clustered with CD-HIT 4.6.5 (Fu et al., 2012) at 40% sequence identity. This threshold was used because flagellar proteins encoded in the SAGs showed sequence identity values ranging from 39% to over 60%. A large cluster containing 1264 protein sequences together with the FlhA sequence of AB-751-O23 was again filtered for nearly full-length sequences (>650 amino acids). The resulting 667 sequences were aligned to the data set used for FlhA/SctV phylogenetic analysis, and the affiliation of the metagenome-derived sequences was determined with Neighbor Joining (1000 bootstraps) in ARB (Ludwig et al., 2004).
Data availability
The assemblies and annotations of the three SAGs have been deposited under the accession numbers FLYF01000001-FLYF01000089 (AB-751-O23), FLYO01000001-FLYO01000054 (AG-110-M15) and FLYP01000001-FLYP01000096 (AG-110-P3) at the European Nucleotide Archive (ENA). Additional sequences used in phylogenetic analyses were deposited under accession numbers LT629158-LT629194 at the European Nucleotide Archive (ENA).
Results and discussion
Novel representatives of deeply branching chlamydial clades
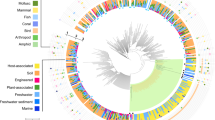
Owing to their host-associated lifestyle, chlamydiae are generally rare in complex microbial communities. Still, we were able to recover three chlamydial SAGs from contrasting marine environments connected to the Pacific and the Atlantic Ocean, respectively. The high-quality SAG assemblies comprised around 1 Mbp each, reflecting around 40% of the predicted genome sizes based on the presence of chlamydial core and single copy genes (Table 1). Phylogenetic analysis of small subunit ribosomal RNA (SSU or 16S rRNA) genes and seven concatenated single-copy marker genes present in all three SAGs demonstrated their affiliation with the phylum Chlamydiae. They represent deeply branching evolutionary lineages within the phylum, clearly distinct from known chlamydiae but related to sequences previously detected in marine samples (Figure 1, Supplementary Figures S1 and S2,Supplementary Text S1) (Pizzetti et al., 2012; Lagkouvardos et al., 2014). Further lines of evidence support their affiliation with chlamydiae: All SAGs encode a high number of chlamydial proteins (~46% show a chlamydial protein as the first blast hit; Table 1), and the respective genes are homogeneously distributed among the contigs (Figure 2). These also include ~42% of the chlamydial core genome, that is, orthologs present in all known chlamydiae (Figure 2). The three SAGs are highly similar with respect to their taxonomic profile (Figure 2) and assignment of genes to functional categories (Supplementary Figure S3), and they share between 179 and 230 pairwise orthologs (Figure 2). Taken together, the three SAGs represent novel, deeply branching marine chlamydiae and provide first insights into their genetic repertoire and biology.

Marine SAGs representing deeply branching chlamydiae. A Bayesian 16S rRNA-based phylogenetic tree is shown, indicating the relationship of the three marine SAGs with known chlamydiae. SAG AG-110-M15 is represented by a partial sequence added to the main tree (dashed line), which included only near full-length sequences. Circles indicate nodes with posterior probabilities >95%. A dendrogram of the tree including branch lengths, posterior probabilities and accession numbers of terminal node sequences is available as Supplementary Figure S1.

Taxonomic profile of chlamydial SAGs and distribution of selected genes. The outer circle (circle 1) illustrates the GC content in a sliding window of 500 nucleotides; for reference, the white line indicates a GC content of 40%. Circle 2 shows the phylum-level taxonomy of the first blast hit of each protein. Highly conserved genes belonging to the chlamydial core genome are indicated in circle 3. The genomic location of flagellar, NF-T3SS and chemotaxis genes is shown in the inner circle. The links represent pairwise orthologs between two SAGs.
Marine chlamydiae with characteristic features of chlamydial biology
All three SAGs include hallmark chlamydial genes (Supplementary Table S1). One of the master regulators of the chlamydial developmental cycle is encoded (Euo, Rosario et al., 2014), which is consistent with microscopical evidence for developmental stages in marine, fish-infecting chlamydiae (Draghi et al., 2004, 2010; Karlsen et al., 2008; Mitchell et al., 2010; Schmidt-Posthaus et al., 2012; Fehr et al., 2013; Stride et al., 2013; Nylund et al., 2015; Steigen et al., 2015; Pawlikowska-Warych and Deptuła, 2016). The major chlamydial virulence mechanism, the type three secretion system (also referred to as non-flagellar type three secretion system, NF-T3SS) (Peters et al., 2007; Stone et al., 2010; Ferrell and Fields, 2016), is represented by structural components, associated chaperones and a number of common chlamydial effector proteins (Ser/Thr protein kinase Pkn5, the macrophage infectivity potentiator Mip; Supplementary Table S1). In addition, a large number of predicted effectors, some of which have the potential to interfere with the host ubiquitination system (Domman et al., 2014) and the highly conserved chlamydia protease-like activity factor CPAF (Grieshaber and Grieshaber, 2014) were identified (Table 1, Supplementary Table S1 and Supplementary Text S2).
Chlamydiae are characterized by reduced metabolic pathways, and although the SAGs represent incomplete genomes, the gene sets found with respect to electron transport chain, amino acid biosynthesis and vitamin biosynthesis resemble those in known chlamydial genomes (Omsland et al., 2014). Notably, each SAG encodes at least two nucleotide transport proteins employed by known chlamydiae to exploit their host cells regarding energy, nucleotides and the cofactor nicotinamide adenine dinucleotide (NAD+) (Tjaden et al., 1999; Haferkamp et al., 2006, 2004; Knab et al., 2011; Fisher et al., 2013; Elwell et al., 2016).
Taken together, the presence of well-conserved chlamydial genes in the three SAGs suggests that these marine chlamydiae show a developmental cycle characteristic for chlamydiae. They use mechanisms for host interaction and exhibit a host-dependent metabolism similar to other members of the phylum (Omsland et al., 2014; Elwell et al., 2016). This is fully consistent with our general notion that the intracellular lifestyle of chlamydiae is ancient and has evolved over a long evolutionary time scale (Greub and Raoult, 2003; Schmitz-Esser et al., 2004; Collingro et al., 2011).
Metabolic adaptations of marine chlamydiae
Yet different chlamydial groups show distinct adaptations (Collingro et al., 2011; Domman et al., 2014; Omsland et al., 2014). In this context, an ABC-transporter for the import of taurine and other sulfonates encoded in one SAG is noteworthy (TauAABC, SCG7086_BT_00100-00070). Taurine is present in high concentrations in most animals including marine invertebrates, molluscs and fish (Huxtable, 1992). Although we could not identify additional genes for taurine usage, taurine could still serve as an energy, carbon, nitrogen or sulfur source for marine chlamydiae (Brüggemann et al., 2004; Denger et al., 2006; Baldock et al., 2007) (Supplementary Text S3).
Most chlamydiae rely on host-derived nucleotides (Tjaden et al., 1999; Haferkamp et al., 2006, 2004; Knab et al., 2011; Fisher et al., 2013; Elwell et al., 2016). Yet, some have the genetic repertoire for pyrimidine de novo synthesis (Bertelli et al., 2015, 2010). This pathway is also complete in one SAG, which in addition contains a nearly full set of genes for purine de novo synthesis (Supplementary Figure S4 and Supplementary Text S4). Thus, this is the first known chlamydia with the potential to thrive completely independent from host-derived nucleotides. However, we found no evidence for the presence of further metabolic pathways, which are generally lacking in other chlamydiae. We thus predict that these marine chlamydiae are still auxotrophic for essential metabolites such as amino acids and cofactors and thus depend on a eukaryotic host.
Flagellar gene sets in marine chlamydiae
The most unexpected finding was the detection of various flagellar genes in addition to genes encoding the NF-T3SS in all three SAGs (20 genes in AB-751-O23 and AG-110-M15, respectively, and 5 genes in AG-110-P3; Figures 2 and Figures 3a). This is highly surprising, because chlamydiae are considered non-motile; there is no evidence that any extant chlamydiae possess flagella or require motility for infectivity. Flagella are composed of components at the cytoplasmic face of the cytoplasmic membrane energizing flagella synthesis and movement, the transmembrane basal body including the motor, and the hook and the (rotating) filament located on the cell surface (Chevance and Hughes, 2008). Few genes with homology to flagellar genes were previously noted in members of the Chlamydiaceae, but those encode exclusively cytoplasmic components, and their role has been cryptic (Peters et al., 2007; Stone et al., 2010; Ferrell and Fields, 2016) (Supplementary Text S5). One SAG (AB-751-O23) is particularly noteworthy, if contigs smaller than 2 kbp are considered in genome reconstruction. With 28 flagellar genes this SAG contains the most complete gene set ever detected in chlamydiae (Figure 3a). The few flagellar genes missing in this SAG are encoded in a syntenic region in one of the other SAGs (AG-110-M15) in which the respective contig spans a larger genomic interval (Figure 3b and Supplementary Text S6). Together this would make up a complete flagellar apparatus including all cytoplasmic, transmembrane and extracellular components (Figure 3a).

Flagellar genes detected in chlamydial SAGs. (a) Schematic overview of components of the flagellar apparatus encoded in three different chlamydial SAGs of marine origin. Notably, they contain orthologs for both the flagellar system and the NF-T3SS (which originally evolved from the flagellar system). All other known chlamydiae lack the majority of flagellar genes. (b) Illustration of the syntenic region of a flagellar gene cluster in two SAGs. Contig ends are indicated.
Flagella as ancient feature of chlamydiae
We investigated the phylogeny of the chlamydial flagellar apparatus using selected proteins encoded on different contigs, including proteins from all three SAGs (Figure 4,Supplementary Table S2 and Supplementary Text S7). None of the chlamydial flagellar proteins showed a close relationship with those of other bacteria, indicating that the chlamydial flagellar apparatus is phylogenetically distinct from recognized flagella. Out of four different data sets analyzed, the SAG flagellar proteins formed a monophyletic group with high support in three trees (Figure 4). In the FlgL tree, only two of the SAGs clustered together (Figure 4d). However, the flagellar hook-associated protein FlgL is located extracellularly and thus shows an elevated evolutionary rate (Nogueira et al., 2012), rendering this particular protein less suitable as a marker to infer evolutionary distant relationships. Together the reconstructed phylogeny of selected flagellar proteins of three chlamydial SAGs favors a scenario in which the flagella of marine chlamydiae share a common evolutionary history. We propose that the flagellar genes of these chlamydiae have not been acquired by (recent) lateral gene transfer, but represent an ancient chlamydial trait that was lost in other known chlamydial lineages.

Common origin of flagellar proteins of marine chlamydiae. Bayesian inference of the phylogeny of selected flagellar proteins encoded at different genomic loci in the chlamydial SAGs. Phylogenetic trees obtained with a concatenated protein data set (a) as well as with individual proteins (b–e) are shown. Homologs of flagellar proteins in the NF-T3SS (SctRSTV) were included if possible. Dotted lines represent branches that have been shortened to enhance clarity. The monophyly of flagellar proteins is well supported in all trees (posterior probability=1), suggesting they are derived from a common ancestor and represent an ancient chlamydial trait. The only exception is the FlgL tree (d), which however is in general inconsistent with the 16S rRNA-based phylogeny of the organisms included (note that other coherent taxonomic groups such as the Proteobacteria or the Firmicutes are also not monophlyetic in this analysis). The original tree files and maximum likelihood phylogenies are available as Supplementary Data 1.
Our analysis included the cryptic flagellar biosynthesis protein FlhA also found in the Chlamydiaceae, which otherwise lack almost all other flagellar genes (Figure 3a and Figure 4b). In the FlhA tree, the Chlamydiaceae proteins formed a monophyletic group, which, however, was markedly divergent from those of flagellated bacteria. This indicates that the Chlamydiaceae FlhA functions in a different context and might rather be used for non-flagellar protein secretion as part of the NF-T3SS. This would be consistent with a current model, suggesting that in Chlamydiaceae flagellar proteins serve to build an alternative NF-T3SS at certain stages in the developmental cycle (Peters et al., 2007; Stone et al., 2010; Ferrell and Fields, 2016). Notably, in this analysis the SAG FlhA protein is most closely related to the FlhA proteins of flagellated free-living bacteria, and well separated from those of the Chlamydiaceae (Figure 4b). This strongly suggests that the SAG FlhA protein is not involved in NF-T3S but indeed functions in a flagellar apparatus.
Chemotaxis systems in marine chlamydiae
In free-living bacteria flagellar motility is regulated by chemotaxis systems, which include transmembrane receptors sensing environmental cues and a two component system controlling flagellar rotation (Wuichet and Zhulin, 2010). All three SAGs contain genes involved in the chemotaxis system (Figure 2), with two SAGs only lacking two genes involved in intracellular signaling cascades (the methyltransferase CheR and the methylesterase CheB; Supplementary Figure S5). A complete chemotaxis gene set has been previously reported in Parachlamydia acanthamoebae (Collingro et al., 2011). In addition, we also identified full gene complements in the environmental chlamydiae Protochlamydia naegleriophila, Criblamydia sequanensis and Estrella lausannensis (Supplementary Figure S5) (Bertelli et al., 2015, 2014). These chlamydiae lack flagellar genes, and it was thus proposed that the chemotaxis genes represent a chemosensory system involved in the regulation of yet unknown cellular functions (Collingro et al., 2011). As complete chemotaxis systems are present in other chlamydiae, and because CheR and CheB are not necessary in all chemotaxis systems (Wuichet and Zhulin, 2010), it seems highly likely that the chemotaxis systems in the marine chlamydiae represented by the SAGs are in fact complete.
Occurrence of chlamydial flagellar genes in the environment
In an attempt to assess the occurrence of flagellar genes of chlamydiae in diverse environments, we searched published metagenomic data using the SAG FlhA sequence as seed. Out of 1200 metagenomic sequences with similarity to the SAG FlhA, 30 represented a near-full-length protein and clustered with the SAG FlhA (Supplementary Table S4). These metagenomic sequences share between 45% and 100% amino acid sequence identity and originate mainly from the Saanich Inlet water column (n=22), where one of the SAGs was isolated (Hawley et al., 2014; Roux et al., 2014). Notably, FlhA sequences were detected in metagenomes from different time points (June 2009 to 2014) and various depths (ranging from 10 to 200 m), suggesting that flagella-encoding chlamydiae are permanently present in this environment (Supplementary Table S4). One sequence was recovered from the northeastern subarctic Pacific Ocean, and others were found in freshwater, soil and bioreactor samples (Supplementary Table S4). As members of the rare biosphere, chlamydiae are poorly represented in metagenomic data sets. The low number of chlamydial FlhA genes recovered is thus not surprising. Nevertheless, this analysis suggests that chlamydiae-encoding flagella are phylogenetically diverse and occur in various, mainly aquatic environments.
Conclusions
In conclusion, this study presents first insights into the biology of deeply branching marine chlamydiae found in geographically distant regions. They show characteristic features of known chlamydial pathogens and symbionts, supporting a scenario in which the intracellular lifestyle and the biphasic developmental cycle were already established in the last common chlamydial ancestor (Greub and Raoult, 2003; Horn et al., 2004; Collingro et al., 2011). Chlamydiae have generally lost redundant genes and non-essential pathways (Nunes and Gomes, 2014; Omsland et al., 2014). The detection of genes encoding flagella thus strongly suggests that this gene set is indeed required and an important component of the biology of marine chlamydiae. Equipped with a flagellar apparatus and a chemotaxis system, marine chlamydiae could be motile and would be able to move actively towards their hosts. This ability might be important in an environment characterized by a low host density and by steady small-scale turbulence and mixing. A conceivable scenario is that flagella are present at the extracellular stage, and that motility of elementary bodies is driven by glycogen as an energy source (Sixt et al., 2013; Gehre et al., 2016). Although the extracellular stage of chlamydiae was traditionally seen as metabolically inert, recent evidence suggest that a limited metabolic activity is maintained in a host-free environment (Haider et al., 2010; Sixt et al., 2013; Omsland et al., 2014, 2012). Whether this is particularly pronounced in marine chlamydiae remains to be explored. In addition, flagella might also facilitate surface attachment to particles in the marine environment. Further, flagella have been implicated in various aspects of bacterial pathogenesis; they are used for adhesion, for host cell entry, as sensors and secretion systems (Chaban et al., 2015).
Our phylogenetic analysis suggests that motility is an ancient chlamydial trait that was subject to differential loss in many known chlamydial lineages. Yet, we provide evidence that chlamydiae with flagella might not be an odd exception, but that flagella-encoding chlamydiae are diverse and occur in different environments. Extant chlamydiae with flagella thus represent a remarkable variation of chlamydial biology, demonstrating that chlamydiae are functionally much more diverse than recognized currently, with potential implications on the structure of trophic interactions in marine ecosystems. Additional research is required to investigate the biology of the many and diverse unknown lineages of this unique bacterial group.
References
Abby SS, Rocha EPC (2012). The non-flagellar type III secretion system evolved from the bacterial flagellum and diversified into host-cell adapted systems. PLoS Genet 8: e1002983.
Abdelrahman YM, Belland RJ (2005). The chlamydial developmental cycle. FEMS Microbiol Rev 29: 949–959.
Baldock MI, Denger K, Smits THM, Cook AM (2007). Roseovarius sp. strain 217: aerobic taurine dissimilation via acetate kinase and acetate-CoA ligase. FEMS Microbiol Lett 271: 202–206.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS et al (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477.
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J et al (2013). GenBank. Nucleic Acids Res 41: D36–D42.
Bertelli C, Aeby S, Chassot B, Clulow J, Hilfiker O, Rappo S et al (2015). Sequencing and characterizing the genome of Estrella lausannensis as an undergraduate project: training students and biological insights. Front Microbiol 6: 101.
Bertelli C, Collyn F, Croxatto A, Rückert C, Polkinghorne A, Kebbi-Beghdadi C et al (2010). The Waddlia genome: a window into chlamydial biology. PLoS One 5: e10890.
Bertelli C, Goesmann A, Greub G (2014). Criblamydia sequanensis harbors a megaplasmid encoding arsenite resistance. Genome Announc 2: 5 e00949–14.
Bou Khalil JY, Benamar S, Baudoin J-P, Croce O, Blanc-Tailleur C, Pagnier I et al (2016). Developmental cycle and genome analysis of ‘Rubidus massiliensis,’ a new Vermamoeba vermiformis pathogen. Front Cell Infect Microbiol 6: 31.
Brüggemann C, Denger K, Cook AM, Ruff J (2004). Enzymes and genes of taurine and isethionate dissimilation in Paracoccus denitrificans. Microbiol Read Engl 150: 805–816.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K et al (2009). BLAST+: architecture and applications. BMC Bioinformatics 10: 421.
Chaban B, Hughes HV, Beeby M (2015). The flagellum in bacterial pathogens: for motility and a whole lot more. Semin Cell Dev Biol 46: 91–103.
Chevance FFV, Hughes KT (2008). Coordinating assembly of a bacterial macromolecular machine. Nat Rev Microbiol 6: 455–465.
Cleland D, Krader P, McCree C, Tang J, Emerson D (2004). Glycine betaine as a cryoprotectant for prokaryotes. J Microbiol Methods 58: 31–38.
Collingro A, Tischler P, Weinmaier T, Penz T, Heinz E, Brunham RC et al (2011). Unity in variety—the pan-genome of the Chlamydiae. Mol Biol Evol 28: 3253–3270.
Dal Conte I, Mistrangelo M, Cariti C, Chiriotto M, Lucchini A, Vigna M et al (2014). Lymphogranuloma venereum: an old, forgotten re-emerging systemic disease. Panminerva Med 56: 73–83.
Darville T (2013). Recognition and treatment of chlamydial infections from birth to adolescence. Adv Exp Med Biol 764: 109–122.
Denger K, Smits THM, Cook AM (2006). Genome-enabled analysis of the utilization of taurine as sole source of carbon or of nitrogen by Rhodobacter sphaeroides 2.4.1. Microbiol Read Engl 152: 3197–3206.
Domman D, Collingro A, Lagkouvardos I, Gehre L, Weinmaier T, Rattei T et al (2014). Massive expansion of ubiquitination-related gene families within the Chlamydiae. Mol Biol Evol 31: 2890–2904.
Draghi A, Bebak J, Daniels S, Tulman ER, Geary SJ, West AB et al (2010). Identification of ‘Candidatus Piscichlamydia salmonis’ in Arctic charr Salvelinus alpinus during a survey of charr production facilities in North America. Dis Aquat Organ 89: 39–49.
Draghi A, Popov VL, Kahl MM, Stanton JB, Brown CC, Tsongalis GJ et al (2004). Characterization of ‘Candidatus piscichlamydia salmonis’ (order Chlamydiales), a chlamydia-like bacterium associated with epitheliocystis in farmed Atlantic salmon (Salmo salar). J Clin Microbiol 42: 5286–5297.
Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797.
Eichinger V, Nussbaumer T, Platzer A, Jehl M-A, Arnold R, Rattei T (2016). EffectiveDB-updates and novel features for a better annotation of bacterial secreted proteins and type III, IV, VI secretion systems. Nucleic Acids Res 44: D669–D674.
Elwell C, Mirrashidi K, Engel J (2016). Chlamydia cell biology and pathogenesis. Nat Rev Microbiol 14: 385–400.
Fehr A, Walther E, Schmidt-Posthaus H, Nufer L, Wilson A, Svercel M et al (2013). Candidatus Syngnamydia venezia, a novel member of the phylum Chlamydiae from the broad nosed pipefish, Syngnathus typhle. PLoS One 8: e70853.
Ferrell JC, Fields KA (2016). A working model for the type III secretion mechanism in Chlamydia. Microbes Infect Inst Pasteur 18: 84–92.
Fisher DJ, Fernández RE, Maurelli AT (2013). Chlamydia trachomatis transports NAD via the Npt1 ATP/ADP translocase. J Bacteriol 195: 3381–3386.
Fu L, Niu B, Zhu Z, Wu S, Li W (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinforma Oxf Engl 28: 3150–3152.
Gehre L, Gorgette O, Perrinet S, Prevost M-C, Ducatez M, Giebel AM et al (2016). Sequestration of host metabolism by an intracellular pathogen. eLife 5: e12552.
Gordon SV, Bottai D, Simeone R, Stinear TP, Brosch R . (2009). Pathogenicity in the tubercle bacillus: molecular and evolutionary determinants. BioEssays News Rev Mol Cell Dev Biol 31: 378–388.
Greub G, Raoult D (2003). History of the ADP/ATP-translocase-encoding gene, a parasitism gene transferred from a Chlamydiales ancestor to plants 1 billion years ago. Appl Environ Microbiol 69: 5530–5535.
Grieshaber SS, Grieshaber NA (2014). The role of the chlamydial effector CPAF in the induction of genomic instability. Pathog Dis 72: 5–6.
Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013). QUAST: quality assessment tool for genome assemblies. Bioinforma Oxf Engl 29: 1072–1075.
Guy L, Kultima JR, Andersson SGE (2010). genoPlotR: comparative gene and genome visualization in R. Bioinforma Oxf Engl 26: 2334–2335.
Haferkamp I, Schmitz-Esser S, Linka N, Urbany C, Collingro A, Wagner M et al (2004). A candidate NAD+ transporter in an intracellular bacterial symbiont related to Chlamydiae. Nature 432: 622–625.
Haferkamp I, Schmitz-Esser S, Wagner M, Neigel N, Horn M, Neuhaus HE (2006). Tapping the nucleotide pool of the host: novel nucleotide carrier proteins of Protochlamydia amoebophila. Mol Microbiol 60: 1534–1545.
Haider S, Wagner M, Schmid MC, Sixt BS, Christian JG, Häcker G et al (2010). Raman microspectroscopy reveals long-term extracellular activity of Chlamydiae. Mol Microbiol 77: 687–700.
Hawley AK, Brewer HM, Norbeck AD, Paša-Tolić L, Hallam SJ (2014). Metaproteomics reveals differential modes of metabolic coupling among ubiquitous oxygen minimum zone microbes. Proc Natl Acad Sci USA 111: 11395–11400.
Horn M (2008). Chlamydiae as symbionts in eukaryotes. Annu Rev Microbiol 62: 113–131.
Horn M, Collingro A, Schmitz-Esser S, Beier CL, Purkhold U, Fartmann B et al (2004). Illuminating the evolutionary history of chlamydiae. Science 304: 728–730.
Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC et al (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44: D286–D293.
Huxtable RJ (1992). Physiological actions of taurine. Physiol Rev 72: 101–163.
Karlsen M, Nylund A, Watanabe K, Helvik JV, Nylund S, Plarre H (2008). Characterization of ‘Candidatus Clavochlamydia salmonicola’: an intracellular bacterium infecting salmonid fish. Environ Microbiol 10: 208–218.
Khodr A, Kay E, Gomez-Valero L, Ginevra C, Doublet P, Buchrieser C et al (2016). Molecular epidemiology, phylogeny and evolution of Legionella. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis 43: 108–122.
Knab S, Mushak TM, Schmitz-Esser S, Horn M, Haferkamp I (2011). Nucleotide parasitism by Simkania negevensis (Chlamydiae). J Bacteriol 193: 225–235.
Knittler MR, Sachse K (2015). Chlamydia psittaci: update on an underestimated zoonotic agent. Pathog Dis 73: 1–15.
Krzywinski MI, Schein JE, Birol I, Connors J, Gascoyne R, Horsman D et al (2009). Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645.
Lagkouvardos I, Weinmaier T, Lauro FM, Cavicchioli R, Rattei T, Horn M (2014). Integrating metagenomic and amplicon databases to resolve the phylogenetic and ecological diversity of the Chlamydiae. ISME J 8: 115–125.
Letunic I, Bork P (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44: W242–W245.
Li L, Stoeckert CJ, Roos DS (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13: 2178–2189.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
Markowitz VM, Chen I-MA, Chu K, Szeto E, Palaniappan K, Grechkin Y et al (2012). IMG/M: the integrated metagenome data management and comparative analysis system. Nucleic Acids Res 40: D123–D129.
Maury MM, Tsai Y-H, Charlier C, Touchon M, Chenal-Francisque V, Leclercq A et al (2016). Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nat Genet 48: 308–313.
Miller MA, Pfeiffer W, Schwartz T (2010). Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proc Gatew Comput Environ Workshop GCE, pp 1–8..
Mitchell SO, Steinum T, Rodger H, Holland C, Falk K, Colquhoun DJ (2010). Epitheliocystis in Atlantic salmon, Salmo salar L., farmed in fresh water in Ireland is associated with ‘Candidatus Clavochlamydia salmonicola’ infection. J Fish Dis 33: 665–673.
Mußmann M, Pjevac P, Krüger K, Dyksma S (2017). Genomic repertoire of the Woeseiaceae/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments. ISME J 11: 1276–1281.
Muyzer G, Smalla K (1998). Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Van Leeuwenhoek 73: 127–141.
Muyzer G, de Waal EC, Uitterlinden AG (1993). Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16 S rRNA. Appl Environ Microbiol 59: 695–700.
Nogueira T, Touchon M, Rocha EPC (2012). Rapid evolution of the sequences and gene repertoires of secreted proteins in bacteria. PLoS One 7: e49403.
Nunes A, Gomes JP (2014). Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect Genet Evol 23: 49–64.
Nylund S, Steigen A, Karlsbakk E, Plarre H, Andersen L, Karlsen M et al (2015). Characterization of ‘Candidatus Syngnamydia salmonis’ (Chlamydiales, Simkaniaceae), a bacterium associated with epitheliocystis in Atlantic salmon (Salmo salar L.). Arch Microbiol 197: 17–25.
Omsland A, Sager J, Nair V, Sturdevant DE, Hackstadt T (2012). Developmental stage-specific metabolic and transcriptional activity of Chlamydia trachomatis in an axenic medium. Proc Natl Acad Sci USA 109: 19781–19785.
Omsland A, Sixt BS, Horn M, Hackstadt T (2014). Chlamydial metabolism revisited: interspecies metabolic variability and developmental stage-specific physiologic activities. FEMS Microbiol Rev 38: 779–801.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25: 1043–1055.
Pawlikowska-Warych M, Deptuła W (2016). Characteristics of chlamydia-like organisms pathogenic to fish. J Appl Genet 57: 135–141.
Peters J, Wilson DP, Myers G, Timms P, Bavoil PM (2007). Type III secretion à la Chlamydia. Trends Microbiol 15: 241–251.
Pizzetti I, Fazi S, Fuchs BM, Amann R (2012). High abundance of novel environmental chlamydiae in a Tyrrhenian coastal lake (Lago di Paola, Italy). Environ Microbiol Rep 4: 446–452.
Pizzetti I, Schulz F, Tyml T, Fuchs BM, Amann R, Horn M et al (2015). Chlamydial seasonal dynamics and isolation of ‘Candidatus Neptunochlamydia vexilliferae’ from a Tyrrhenian coastal lake. Environ Microbiol 18: 2405–2417.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P et al (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41: D590–D596.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng J-F et al (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437.
Robins WP, Mekalanos JJ (2014). Genomic science in understanding cholera outbreaks and evolution of Vibrio cholerae as a human pathogen. Curr Top Microbiol Immunol 379: 211–229.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S et al (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61: 539–542.
Rosario CJ, Hanson BR, Tan M (2014). The transcriptional repressor EUO regulates both subsets of Chlamydia late genes. Mol Microbiol 94: 888–897.
Roulis E, Polkinghorne A, Timms P (2013). Chlamydia pneumoniae: modern insights into an ancient pathogen. Trends Microbiol 21: 120–128.
Roux S, Hawley AK, Torres Beltran M, Scofield M, Schwientek P, Stepanauskas R et al (2014). Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta-genomics. eLife 3: e03125.
Schmidt-Posthaus H, Polkinghorne A, Nufer L, Schifferli A, Zimmermann DR, Segner H et al (2012). A natural freshwater origin for two chlamydial species, Candidatus Piscichlamydia salmonis and Candidatus Clavochlamydia salmonicola, causing mixed infections in wild brown trout (Salmo trutta). Environ Microbiol 14: 2048–2057.
Schmitz-Esser S, Linka N, Collingro A, Beier CL, Neuhaus HE, Wagner M et al (2004). ATP/ADP translocases: a common feature of obligate intracellular amoebal symbionts related to Chlamydiae and Rickettsiae. J Bacteriol 186: 683–691.
Sixt BS, Siegl A, Müller C, Watzka M, Wultsch A, Tziotis D et al (2013). Metabolic features of Protochlamydia amoebophila elementary bodies—a link between activity and infectivity in Chlamydiae. PLoS Pathog 9: e1003553.
Stamatakis A . (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinforma Oxf Engl 30: 1312–1313.
Steigen A, Karlsbakk E, Plarre H, Watanabe K, Øvergård A-C, Brevik Ø et al (2015). A new intracellular bacterium, Candidatus Similichlamydia labri sp. nov. (Chlamydiaceae) producing epitheliocysts in ballan wrasse, Labrus bergylta (Pisces, Labridae). Arch Microbiol 197: 311–318.
Stepanauskas R (2012). Single cell genomics: an individual look at microbes. Curr Opin Microbiol 15: 613–620.
Stone CB, Bulir DC, Gilchrist JD, Toor RK, Mahony JB . (2010). Interactions between flagellar and type III secretion proteins in Chlamydia pneumoniae. BMC Microbiol 10: 18.
Stride MC, Polkinghorne A, Miller TL, Groff JM, Lapatra SE, Nowak BF (2013). Molecular characterization of ‘Candidatus Parilichlamydia carangidicola,’ a novel Chlamydia-like epitheliocystis agent in yellowtail kingfish, Seriola lalandi (Valenciennes), and the proposal of a new family, ‘Candidatus Parilichlamydiaceae’ fam. nov. (order Chlamydiales). Appl Environ Microbiol 79: 1590–1597.
Swan BK, Martinez-Garcia M, Preston CM, Sczyrba A, Woyke T, Lamy D et al (2011). Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean. Science 333: 1296–1300.
Taylor-Brown A, Vaughan L, Greub G, Timms P, Polkinghorne A (2015). Twenty years of research into Chlamydia-like organisms: a revolution in our understanding of the biology and pathogenicity of members of the phylum Chlamydiae. Pathog Dis 73: 1–15.
Tjaden J, Winkler HH, Schwöppe C, Van Der Laan M, Möhlmann T, Neuhaus HE (1999). Two nucleotide transport proteins in Chlamydia trachomatis, one for net nucleoside triphosphate uptake and the other for transport of energy. J Bacteriol 181: 1196–1202.
Vanthournout B, Hendrickx F (2015). Endosymbiont dominated bacterial communities in a dwarf spider. PLoS One 10: e0117297.
Wagner M, Horn M (2006). The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr Opin Biotechnol 17: 241–249.
Weinmaier T, Platzer A, Frank J, Hellinger H-J, Tischler P, Rattei T (2016). ConsPred—a rule-based (re-)annotation framework for prokaryotic genomes. Bioinforma Oxf Engl 32: 3327–3329.
Wright HR, Turner A, Taylor HR (2008). Trachoma. Lancet Lond Engl 371: 1945–1954.
Wuichet K, Zhulin IB (2010). Origins and diversification of a complex signal transduction system in prokaryotes. Sci Signal 3: ra50.
Zaikova E, Walsh DA, Stilwell CP, Mohn WW, Tortell PD, Hallam SJ (2010). Microbial community dynamics in a seasonally anoxic fjord: Saanich Inlet, British Columbia. Environ Microbiol 12: 172–191.
Acknowledgements
We thank Florian Goldenberg and Thomas Rattei (Division of Computational Systems Biology, University of Vienna) for providing and maintaining the Life Science Compute Cluster (LiSC) at the University of Vienna, and Thomas Weinmaier and Alexander Platzer for help with the ConsPred annotation pipeline. We are grateful to Petra Pjevac for assistance in data analysis. Staff at the MPI Bremen, the MP-GC Cologne and the Bigelow Laboratory Single Cell Genomics Center are acknowledged for sample preparation and sequencing. This work was supported by a European Research Council Grant (ERC StG EVOCHLAMY, no. 281633) and the Austrian Science Fund FWF (Grant I291-B09) to MH. RS was supported by the US National Science Foundation grants DEB-1441717 and OCE-1335810. SH was supported by the G Unger Vetlesen and Ambrose Monell Foundations, the Tula Foundation funded Centre for Microbial Diversity and Evolution, Natural Sciences and Engineering Research Council (NSERC) of Canada, Canada Foundation for Innovation (CFI) and the Canadian Institute for Advanced Research (CIFAR).
Author contributions
AC and MH conceived the study. MM, SH and RS provided samples and organized single-cell sorting and sequencing. AC, SK and RS analyzed the data. AC and MH wrote the manuscript. All authors commented on and edited the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Collingro, A., Köstlbacher, S., Mussmann, M. et al. Unexpected genomic features in widespread intracellular bacteria: evidence for motility of marine chlamydiae. ISME J 11, 2334–2344 (2017). https://doi.org/10.1038/ismej.2017.95
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.95
This article is cited by
-
Gene gain facilitated endosymbiotic evolution of Chlamydiae
Nature Microbiology (2023)
-
Ecology and evolution of chlamydial symbionts of arthropods
ISME Communications (2022)
-
Genomic diversity and biosynthetic capabilities of sponge-associated chlamydiae
The ISME Journal (2022)
-
Presence of DNA from Chlamydia-like organisms in the nasal cavities of grey seal pups (Halichoerus grypus) and three different substrates present in a breeding colony
BMC Veterinary Research (2021)
-
Pangenomics reveals alternative environmental lifestyles among chlamydiae
Nature Communications (2021)
