
Abstract
The terrestrial deep subsurface is a huge repository of microbial biomass, but in relation to its size and physical heterogeneity, few sites have been investigated in detail. Here, we applied a culture-independent metagenomic approach to characterize the microbial community composition in deep (1500 meters below surface) terrestrial fluids. Samples were collected from a former gold mine in Lead, South Dakota, USA, now Sanford Underground Research Facility (SURF). We reconstructed 74 genomes from metagenomes (MAGs), enabling the identification of common metabolic pathways. Sulfate and nitrate/nitrite reduction were the most common putative energy metabolisms. Complete pathways for autotrophic carbon fixation were found in more than half of the MAGs, with the reductive acetyl-CoA pathway by far the most common. Nearly 40% (29 of 74) of the recovered MAGs belong to bacterial phyla without any cultivated members—microbial dark matter. Three of our MAGs constitute two novel phyla previously only identified in 16 S rRNA gene surveys. The uniqueness of this data set—its physical depth in the terrestrial subsurface, the relative abundance and completeness of microbial dark matter genomes and the overall diversity of this physically deep, dark, community—make it an invaluable addition to our knowledge of deep subsurface microbial ecology.
Similar content being viewed by others


Introduction
Most of the Earth’s deep subsurface biosphere (DSB) is energy-starved and functionally defined by the exclusive presence of microbial life and the lack of light or light-derived biomass. The DSB, in particular the terrestrial component, has only recently been appreciated as dynamic, populated, metabolically active, interacting with and perhaps controlling global elemental cycles. In terrestrial environments, a functional definition mandates that the DSB be independent from photosynthetically derived organic matter and reliant on endogenous sources of energy (Fredrickson and Onstott, 1996; Stevens, 1997). However, it is impossible to know definitively that a subsurface environment is truly independent from surface-derived products without significant study. In this study, we define deep similar to Orcutt et al. (2011) and Lovely and Chapelle (1995): the DSB is an environment absent of photosynthesis and isolated from direct contact with surface waters.
It has also been shown recently that the terrestrial DSB harbors a great abundance and diversity of microorganisms (for example, Chivian et al., 2008; Rinke et al., 2013; Dong et al., 2014; Lau et al., 2014; Nyyssönen et al., 2014; Magnabosco et al., 2015; Baker et al., 2016). Early estimates of terrestrial subsurface cells were on the order of 0.25–2.5 × 1030 (Whitman et al., 1998). More recent estimates put the total deep subsurface biomass at 16–157 Pg C, with the terrestrial part accounting for 14–135 Pg C (Kallmeyer et al., 2012; McMahon and Parnell, 2014). However, the microbial physiologies, corresponding metabolisms and their reaction energetics remain almost completely unmapped.
The carbon sources and cycling processes in the vast terrestrial DSB are of particular interest (McMahon and Parnell, 2014). Due in large part to limited global samples, these sources and processes remain poorly constrained (Onstott et al., 1998; Pfiffner et al., 2006; Simkus et al., 2016). Metagenomic and single cell genomic sequencing studies in shallow (⩽100 m) terrestrial systems provided insight into metabolic capabilities of microbial dark matter (Rinke et al., 2013) and genomic expansion of the domain Archaea (Tyson et al., 2004; Castelle et al., 2015; Youssef et al., 2015a; Baker et al., 2016; Seitz et al., 2016). However, metagenomic analyses of samples from the deeper terrestrial biosphere remain rare (see Edwards et al., 2006; Chivian et al., 2008; Dong et al., 2014; Lau et al., 2014; Nyyssönen et al., 2014; Magnabosco et al., 2015).
In an effort to understand the metabolic capabilities of microbial communities in the terrestrial DSB, we performed random shotgun metagenomic sequencing on whole genomic DNA extracted from two separate fluid samples collected 1.5 kilometers below surface (kmbs). Genomes from the two metagenomes were binned and phylogenomic and 16 S rRNA sequence analyses were used to taxonomically classify them. These curated metagenome assembled genomes (MAGs) were interrogated for metabolic capabilities including electron donor and acceptor usage, and heterotrophic and autotrophic carbon utilization. In addition, genomes from two new candidate phyla were identified. Two genomes, SURF_5 and SURF_17 are the first members of a new candidate phylum, designated SURF-CP-1 and named Abyssubacteria, Latin prefix meaning deep, owing to their collection 1.5 km below surface. One genome, SURF_26, is the first member of a new candidate phylum, initially designated SURF-CP-2 and named Aureabacteria, Latin prefix meaning gold, to represent its collection in the former Homestake gold mine.
Materials and methods
Field sampling
All fluid samples and corresponding geochemical data were collected in the former Homestake gold mine (now Sanford Underground Research Facility, SURF) near Lead, South Dakota, USA (44°21′ N 103°45′ W) in October 2013. Both samples are deep subsurface fracture fluids from legacy boreholes drilled ~1.5 kmbs, and 600 and 900 horizontal feet (180 and 270 m) into host rock. The SURF archive names, given to these boreholes in 2001 at the time of drilling, are DUSEL-B and DUSEL-D, but for the sake of simplicity, we will hereafter refer to the borehole fluid samples as SURF-B and SURF-D, respectively. A comprehensive description of sampling methods for geochemistry can be found in Osburn et al. (2014). Details of samples and sample locations are provided in Table 1.
DNA extraction and sequencing
Total microbial cells were collected from borehole fluids on 47 mm, 0.2 μm Supor filters (Pall Corporation, Port Washington, NY, USA), which were then stored on dry ice, transported to the University of Southern California, and frozen at −80 °C. Whole genomic DNA was extracted using a modified phenol–chloroform method with ethanol precipitation as previously described in Momper et al. (2015). DNA concentration was checked on a Qubit 2.0 fluorometer (Thermo Fisher Scientific, Chino, CA, USA), and purity was measured on a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) before samples were sent for sequencing. Sequencing was performed at the University of Southern California’s Genome and Cytometry Core Facility (Los Angeles, CA, USA). Illumina sequencing libraries were prepared according to Dunham and Friesen (2013) with the exception that DNA was sheared with dsDNA Shearase Plus (Zymo: Irvine, CA, USA) and cleaned using Agencourt AMPure XP beads (Beckman-Coulter: Indianapolis, IN, USA). Fragment size selection was also carried out using beads instead of gel electrophoresis. Libraries were quantified using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific), and the fragment size distribution was determined with an Agilent Bioanalyzer 2100. The libraries were then pooled in equimolar concentrations, quantified via qPCR using the Kapa Biosystems Library Quantification Kit and paired-end sequenced on an Illumina HiSeq 2500 platform. The libraries preparation, pooling, quality control and sequencing were all performed at the University Park Campus Genome Core (University of Southern California, Los Angeles, CA, USA).
De novo assembly and read mapping
Quality control was performed using Trimmomatic 0.36 with default parameter and a minimum sequence length of 36 base pairs (Bolger et al., 2014). Reads were assembled using IDBA-UD v1.1.1 (Peng et al., 2012) with a 5000 bp minimum contig length. Sequences from each of the two borehole fluids were assembled individually, and together as a co-assembly. All downstream analyses reported here were performed on the co-assembly because (a) the co-assembly produced a longer maximum contig length, (b) a larger number of contigs were produced and (c) preliminary 16 S data (Osburn et al., 2014) indicated highly similar community composition between the two fluids, and initial metabolic and phylogenetic analyses from the individual assemblies were producing redundant results (data not shown). Coverage depth information was then generated for scaffolds greater than 5000 base pairs by mapping the 150 base pair paired-end reads of each of the two samples to the co-assembled scaffolds using Bowtie2 v2.2.6 (Langmead and Salzberg, 2012) with the BWA-SAMPLE algorithm and default parameters. SAMtools v0.1.17 (Li et al., 2009) was then used to convert files to binary format for downstream analysis.
Generation of MAGs
MAGs were generated using sequence composition, differential coverage and read-pair linkage through the CONCOCT program within the Anvi’o software (Alneberg et al., 2014; Eren et al., 2015). MAGs were manually refined and curated using an interactive interface in the Anvi’o program (Eren et al., 2015). After refinement, MAG completeness (reported as percentage of the set of single-copy marker genes present) and contamination (calculated as multiple occurrence of a single-copy marker gene) were re-calculated using five different standard marker gene suites (Creevey et al., 2011; Dupont et al., 2012; Wu and Scott, 2012; Campbell et al., 2013; Alneberg et al., 2014) (Supplementary Figure 1).
Assignment of putative taxonomies
MAGs were assigned putative taxonomic identities according to their placement in a phylogenome tree using the ‘tree’ command in CheckM (Parks et al., 2015). CheckM employs pplacer (Matsen et al., 2010) to place concatenated amino-acid alignments into an Integrated Microbial Genomes (IMG) database of complete genomes (CheckM database v1.0.4). Phylogenetic identities of MAGs were further refined according to information from 16 S rRNA and other conserved single-copy marker genes, as described below.
16 S rRNA tree construction
Small subunit ribosomal RNA genes (>300 nucleotides) were extracted from the MAGs using the ‘ssu_finder’ tool integrated within CheckM (Parks et al., 2015) and their three closest neighbors identified via a BLAST (Basic Local Alignment Search Tool) query against the non-redundant NCBI database. All sequences were pooled and aligned using the online SINA tool v1.2.11 (Pruesse et al., 2012). For comparison, additional SSU rRNA sequences from Kantor et al. (2013), Rinke et al. (2013) and Castelle et al. (2015) were aligned in a similar fashion. All aligned sequences were imported ARB v6.0.3 (Ludwig et al., 2004), and additional closest relatives to the MAG SSU rRNA genes were identified within the SSURef_NR99_123_SILVA_12_07_15 and LTPs123_SSU databases (Pruesse et al., 2007; Yarza et al., 2008; Quast et al., 2013). A maximum-likelihood phylogenetic analysis was performed using RAxML v8.2.8 with the GTR model of nucleotide substitution under the gamma—and invariable—models of rate heterogeneity (Stamatakis, 2006).
Phylogenomic analyses
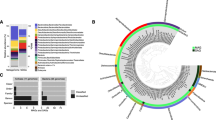
From all SURF MAGs described here with completeness >50% and relevant MAGs and SAGs (single-amplified genomes) from IMG (Markowitz et al., 2014), ggKbase and National Center for Biotechnology Information (NCBI) GenBank databases, phylogenetically-informative marker genes were identified and extracted using the ‘tree’ command in CheckM. In CheckM, open reading frames were called using prodigal v2.6.1 (Hyatt et al., 2012) and a set of 43 lineage-specific marker genes, similar to the universal set used by PhyloSift (Darling et al., 2014), were identified and aligned using HMMER v3.1b1 (Eddy, 2011). The 75 MAGs with>50% completeness were given taxonomic identifications through analysis of a concatenated marker gene alignment (6988 amino-acid positions) and placement in a phylogenomic tree with closest related MAGs and SAGs found in the NCBI, IMG and ggKbase databases. The phylogeny was produced using FastTree v2.1.9 (Price et al., 2010) with the WAG amino-acid substitution model and ‘fastest’ mode and bootstrap values reported by FastTree analysis indicate local support values (Figure 1).

Diversity of organisms from which genomes from metagenomes (MAGs) were reconstructed from the Sanford Underground Research Facility fluids. Maximum-likelihood phylogenomic analysis of all MAGs >50% complete and select genomes, MAGs and single cell amplified genomes from IMG, NCBI and ggKbase. The scale bar corresponds to 1 substitution per amino-acid position. Each numbered and blue-colored branch represents one MAG identified here. The names of major lineages with MAGs found in SURF fluids are indicated with bold-face font. MAGs belonging to novel phyla for which no reference genomes identified (SURF-CP-1 and SURF-CP-2) are indicated. Black (100%), gray (⩾75%) and white (⩾50%) circles indicate nodes with high local support values, from 1000 replicates.
Metabolic pathway analysis
Co-assembled metagenomes and individual genomes were submitted for gene calling and annotations through the DOE Joint Genome IMG-MER (Institute Integrated Microbial Genomes metagenomics expert review) pipeline (Markowitz et al., 2008; Huntemann et al., 2015). Genes of encoding metabolic and other functions of particular interest were queried in IMG-MER and associated to the previously described MAGs using common scaffold ID identifiers. Functional genes that were found in candidate phyla bins in this study that had not been reported previously in those phyla were scrutinized using additional methods; specifically, the coding region for each gene of interest was extracted, translated and used to perform a BLASTp search for nearest neighbors. Alignments were examined and, if the alignment was of poor quality (for example, large gaps and/or low identity), the gene was deemed a false hit and was not included in our results and discussion. A complete list of metabolic genes of interest that were queried in this study can be found in Supplementary Table 1.
Possible autotrophy was investigated in all MAGs. We examined KEGG (Kyoto Encyclopedia for Genes and Genomes) biochemical maps for the six known carbon fixation pathways in each MAG, but only genes that are known to code for enzymes unique to carbon fixation were included (for example, genes involved in glycolysis were not included in the gene suite for the reductive citric acid cycle). A complete list of the KEGG identifiers for each of the six pathways can be found in Supplementary Table 2.
Results
Sequencing and assembly
Shotgun sequencing of total community genomic DNA produced 147 742 812 and 137 946 268 150 base pair (bp) paired-end reads for SURF-B and –D fluids, respectively. After quality filtering, 94.68% of reads were retained for assembly. De novo assemblies of quality-filtered reads generated a total of 637 833 contigs for the co-assembly. Maximum contig length was 576 430 bp. Prediction of open reading frames resulted in 1 187 179 putative genes in the co-assembly (Table 1).
MAGs
A total of 74 MAGs with >50% completeness and <10% contamination were recovered from the co-assembled metagenomes. Genome statistics including number of scaffolds, genes, genome size, average completeness and contamination are listed in Table 2. Bins were assigned numerical identifiers in order of decreasing completeness. Of the 74 individual MAGs, 22 were >90% complete and 15 were 80–90% complete. Completeness and contamination was averaged from five sets of widely accepted single-copy marker genes (Supplementary Figure 1; Creevey et al., 2011; Dupont et al., 2012; Wu and Scott, 2012; Campbell et al., 2013; Alneberg et al., 2014). Standard deviation of these values is reported in Table 2.
MAG phylogenetic identification
The majority of the SURF MAGs (72 of 74) were from the domain Bacteria; only two were from the domain Archaea, specifically the phylum Woesearchaeota (Figure 1). Within the Bacteria, members of the class Deltaproteobacteria are highly represented (16 MAGs) in both SURF-B and –D fluids. Recruitment of the MAGs found in SURF fluid indicates similar coverage values for most genomes investigated (data not shown). The exceptions included numerous members (SURF_49, 50, 63, 73) of the Patescibacteria superphylum, Microgenomates (formerly OP11) and Parcubacteria (formerly OD1), which have relatively higher coverage in SURF-D fluids. We note that the MAGs for these two phyla are 50–75% complete and comprise many of our MAGs that are <80% complete (Table 2, Figure 2 and Supplementary Figure 1).

Functional genes, genome size, GC content and small subunit 16S rRNA presence in MAGs. Heat maps indicate total MAG scaffold size, number of contigs, GC content, completeness and contamination. Presence and absence of all SSU rRNA genes >300 bp and presence of genes encoding functional genes are indicated by black boxes: pmoA, particulate methane monooxygenase; nrfAD, nitrite reductase; norBC, nitric oxide reductase; nosZDFY, nitrous oxide reductase; dsrAB, dissimilatory sulfite reductase; napABC, periplasmic nitrate reductase; narABDG, nitrate reductase; nirBDG, nitrite reductase; hydB, sulfur reductase; phsA, thiosulfate reductase; redH, reductive dehalogenase; ICL, isocitrate lyase.
Candidate phyla make up almost 40% (29 of 74) of the MAGs in deep fluids at SURF. MAGs belonging to the bacterial candidate phyla Zixibacteria (formerly RBG-1), Omnitrophica (formerly OP3), WCHB1-60, Parcubacteria (formerly OD1), Microgenomates (formerly OP11), WWE3 and Latescibacteria (formerly WS3) were recovered from SURF fluids. One MAG (SURF_60) is most closely related to candidate phylum Candidatus Desulforudis audaxviator, a member of the phylum Firmicutes that has been found globally in deep subsurface environments (Baker et al., 2003; Cowen et al., 2003; Chivian et al., 2008; Aüllo et al., 2013; Tiago and Veríssimo, 2013; Magnobosco et al., 2015; Jungbluth et al., 2016). At last, two of our MAGs (SURF_58 and SURF_65) are affiliated with the recently named archaeal candidate phylum Woesearchaeota (Castelle et al., 2015).
Our 16 S rRNA gene phylogenetic analysis showed that four MAGs (SURF_12, 18, 25 and 26) are related to the Omnitrophica/OP3, but they were polyphyletic relative to the OP3/Omnitrophica group (Supplementary Figure 2). In-depth phylogenomic analysis using concatenated ribosomal proteins of publicly available genomes from both SAGs and MAGs revealed a distinction between the Omnitrophica and OP3, and the existence of two phyla, not one (Figure 1 and Supplementary Figure 2). MAGs SURF_12, 18, 25 and 26 were further investigated by a BLAST search of all coding regions within the genomes against all publicly available genomes (BLAST2GO, v1.3, BioBam, Valencia Spain). Results revealed that MAGs SURF_12 is a member of the candidate phylum Omnitrophica; MAGs SURF_18 and SURF_25 are members of the candidate phylum OP3 (Supplementary Figure 3), phyla that were previously grouped together as a single phylum, but with inclusion of new additional data appear to be two distinct phyla. MAG SURF_26 is the first member of a new candidate phylum, here named SURF-CP-2 (Figure 1 and Supplementary Figure 4). Similar phylogenetic classification obstacles were encountered with genomes SURF_5 and SURF_17. Genes were translated into amino-acid sequences and phylogenomic analysis of concatenated marker genes and single-copy marker genes and review of top species hits results indicate that these MAGs constitute the first members of a novel candidate phylum, here designated SURF-CP-1, which is phylogenetically related to the Omnitrophica and Planctomycetes (Figure 1 and Supplementary Figure 5).
Metabolic capabilities in MAGs: inferred electron donors
Presence of functional genes was queried in MAGs and all results can be found in Figure 2. Particulate methane (pmoA) monooxygenase was identified in candidate phylum Omnitrophica MAG SURF_12 only. To our knowledge this is the first report of putative methanotrophic capability in the candidate phylum Omnitrophica. Nickel-Iron (Ni-Fe) and (Fe-Fe) hydrogenases were present in >10% of all MAGs (9 of 74), possibly indicating a widespread ability to utilize hydrogen as an electron donor. Homologs of formate dehydrogenase (fdhABC) were queried in all genomes, but no single genome contained genes for all three subunits (ABC) for this multimeric protein (Figure 2). Similarly, genes involved in carbon monoxide oxidation (coxMLS) were searched for but only homologs for coxS were identified (Supplementary Table 1). Canonical genes involved in thiosulfate, sulfur or sulfide oxidation, respectively (soxBCY, sor, sqr, fcc) were queried in the MAGs as well as in all of the scaffolds from both metagenomes, but none were found. Genes indicative of sulfur oxidation via the reverse dissimilatory sulfate reduction pathway (dsrEFH) (Ghosh and Dam, 2009) were found in three genomes that also contained a homolog for dissimilatory sulfite reductase (dsrAB). However, dsrL, which is considered the essential enzyme for sulfur oxidation in this pathway (Sander et al., 2006; Grimm et al., 2008; Ghosh and Dam, 2009) is not present in any genomes (Figure 2). Genes encoding enzymes involved in ferrous iron oxidation (Supplementary Table 1) were not found in any genome recovered from these fluids.
Metabolic capabilities in MAGs: inferred electron acceptors
Putative sulfate/sulfite reducing microorganisms are relatively abundant among the 74 reconstructed MAGs in this study, with 20% (13 of 74, Figure 2) containing the genes for dissimilatory sulfite reductase (dsrAB) and the necessary accessory protein dsrD. The genes encoding for cytoplasmic nitrate reductase (nar, all enzyme subunits) were identified in 35% (27 of 74) of the MAGs. All subunits of the nar operon (ABDG) were found in MAG SURF_12, belonging to the candidate phylum Omnitrophica. Conversely, putative periplasmic nitrate reduction ability (napABC) was less common, found in only seven MAGs. Nitrite reductase (nirBDG) and nitric oxide reductase (norBC) were present in 10 and 19 MAGs, respectively, but nitrous oxide reductase (nosZDFY) was only found in three MAGs.
It should be noted that genes for enzymes and co-factors involved in methane transformation (mcrA, coenzyme F420) and cellulose degradation (cel5, cel48) were found on scaffolds in the assembled metagenomes but were not found on scaffolds in the MAGs. In addition, genes involved in extracellular iron reduction (mtrA), and tetrahydromethanopterin-linked C1 transfer (fae and fhcD) were queried but not found in MAGs or the full assembled metagenomes as a whole (Supplementary Table 1).
Modes of carbon fixation in MAGs
Carbon fixation capability was examined in each of the 74 MAGs (Figure 3). The reductive Acetyl-CoA (Wood-Ljungdahl) pathway was the most common; 33 MAGs contained at least 75% of the necessary genes involved in this pathway (essential genes are listed in Supplementary Table 2). The corresponding lineages were diverse and included Ammonifex, Ca. Desulforudis, Dehalococcoidia, Dethiobacter, numerous Deltaproteobacteria, Actinobacteria, Firmicutes and Chloroflexi, as well as members of the candidate phyla Omnitrophica and Hydrogenedentes. Only one MAG contained the gene encoding for RuBisCO and phosphoribulokinase, the canonical enzymes involved in carbon fixation via the reductive pentose phosphate (Calvin) cycle. This MAG was a member of the Gammaproteobacteria. The sequences were homologous with known Type II RuBisCO, which catalyzes the carboxylation and oxygenation of ribulose 1,5-bisphosphate (Tabita et al., 2008). The four other carbon fixation pathways (3-hydroxypropionate bi-cycle, 3-hydroxypropionate/4-hydroxybutyrate, dicarboxylate/4-hydroxybutyrate, reductive citric acid cycle) were far less common in MAGs (Figure 3) and in general less complete. No MAG contained all of the known genes involved in any of these pathways, but numerous members of the Deltaproteobacteria contained >80% of the necessary genes for the reductive citric acid, 3-hydroxypropionate bi-cycle, 3-hydroxypropionate/4-hydroxybutyrate and dicarboxylate/4-hydroxybutyrate cycles (Figure 3).

Identification of carbon fixation capabilities in all MAGs >50% complete. Heat map indicates the percent of signature genes present in six well-characterized carbon fixation pathways. A full list of genes queried is described in Supplementary Table 2.
Discussion
Next-generation Illumina sequencing technology has only been used in a few terrestrial deep biosphere studies to explore microbial community composition and metabolic capabilities. Dong et al. (2014) showed that one bacterial species, Halomonas sulfidaeris, dominated the community in a 1.8 km-deep Cambrian Sandstone reservoir. Similarly, Chivian et al. (2008) found a mono-species community in 2.8 km-deep fracture fluids in a South African gold mine. In contrast to the Dong et al. (2014) and Chivian et al. (2008) studies, the present study of fluids from the 1.5 km-deep Paleoproterozoic metasedimentary units at SURF, found a diverse subsurface community residing in the terrestrial DSB. This is more in line with similar studies by Lau et al. (2014), Nyyssönen et al. (2014) and Maganbosco et al. (2015). This highlights the microbial variability, both in terms of cell density and diversity within terrestrial subsurface fluids. In this study, we found similar phylogenetic diversity as that reported in Magnabosco et al (2015) from 3 km-deep Precambrian continental crust in the Witwatersrand Basin of South Africa. In both studies ~25 bacterial phyla were identified in the community. The major difference in phylogeny was the large proportion of candidate phyla identified in this study and almost complete absence of candidate phyla in the other—a mere four groups could only be identified on the domain level (Magnabosco et al., 2015). This study recovered a total of 74 MAGs, 22 of which are high quality genomes, meaning >90% complete and <5% contaminated by recent standards (Bowers et al., 2017). The remaining 52 MAGs are >50% complete and <10% contaminated, deemed medium quality by current standards (Bowers et al., 2017). Here, we discuss trends in metabolic capability among these 74 MAGs from SURF fluids, with a particular focus on subsurface microbial dark matter represented by near-complete candidate phyla genomes. Given the in situ geochemical conditions and calculations of redox reaction energetics (Osburn et al., 2014), particular interest was paid to energy metabolisms involving cycling of hydrogen, nitrogen, sulfur and methane.
Biological transformation of nitrogen, sulfur and hydrogen
Evidence of denitrification (nar, nap, nir, nor, nos and/or nrf) was found in 38 MAGs. The commonality of both cytoplasmic (narABDG) and periplasmic (napABC) nitrate reductases and all other enzymes that perform steps in the complete denitrification (NO3−→N2) process suggests that dissimilatory nitrogen-transforming metabolisms are common and there is likely a dynamic nitrogen cycle occurring within SURF fluids. Note that although nitrite levels were below detection limit, nitrate measured 10.3 and 23.7 μm in SURF-D and –B fluids, respectively at the time these samples were collected (Osburn et al., 2014). Thermodynamic calculations indicate that nitrate reduction (especially with hydrogen as an electron donor) is highly exergonic in SURF-B and –D fluids (Osburn et al., 2014).
Genes for putative hydrogen-oxidizing enzymes (Ni-Fe and Fe-Fe hydrogenases) were common (10 of 74 genomes), however dissolved molecular hydrogen was detected in only nanomolar levels in SURF fluids. Despite the extremely low measured concentrations, hydrogen is still an energetically favorable electron donor for most redox couples considered here (Osburn et al., 2014). The common occurrence of Ni-Fe and Fe-Fe hydrogenases and discrepancy with measured hydrogen could be due to multiple factors. First, these genes may not be expressed and could be turned on opportunistically if hydrogen concentrations increase. Second, the low solubility and rapid escapes of hydrogen make it an extremely difficult gas to measure accurately in situ. Concentrations experienced by microbes in situ may very well be higher than laboratory-measured concentrations. Forthcoming metatranscriptomics should shed light on the active use of hydrogen as an electron donor in these deep fluids.
Putative sulfate reducers are abundant among the MAGs in this study, with ~20% of all bins containing the dsrABD genes (Figure 2). Interestingly, the genes for dsrABD and Ni-Fe, Fe-Fe hydrogenases were present in SURF_60, a phylogenetic relative to Ca. Desulforudis audaxviator, a member of the Firmicutes that has been found in other terrestrial and marine subsurface environments (Baker et al., 2003; Jungbluth et al., 2013; Magnobosco et al., 2015). For example, in fracture water in South Africa it was found to dominate (>99%) the microbial community (Lin et al., 2006; Chivian et al., 2008). Genome analysis of that lineage revealed an almost self-sufficient chemolithoautotrophic bacterium, putatively capable of carbon and nitrogen fixation and sulfate reduction using hydrogen as an electron donor (Chivian et al., 2008).
Note that canonical genes involved in thiosulfate oxidation, sulfur oxidation or sulfide oxidation (soxBCY, sor, sqr, fcc) were not detected in any of our SURF MAGs, nor were they found when all scaffolds in the metagenomes were queried (Supplementary Table 1). This could indicate that sulfur species are rarely, if ever, used as an electron donor in SURF fluids. This may seem counterintuitive given the relatively high total sulfide levels (83–130 μg l−1) in SURF-B and –D fluids (Osburn et al., 2014). However, thermodynamic calculations demonstrate that when considering energy density (J kg−1 H2O), sulfide oxidation with either oxygen or nitrate as the oxidant is not favorable (that is, endergonic) (Osburn et al., 2014). Using a combination of metagenomic, geochemical and thermodynamic data, we conclude that sulfate reduction to elemental sulfur or sulfide is likely an important energy metabolism in these subsurface fluids. However, the oxidation of reduced sulfur back to sulfate appears to be a rare metabolic strategy, most likely because other electron donors (example, methane, carbon monoxide, hydrogen, ferrous iron) have a higher energy density than reduced sulfur species such as elemental sulfur and sulfide (Osburn et al., 2014). This highlights the importance of considering energy density, not only Joules per mole of electrons transferred, when modeling in situ thermodynamic yields of dissimilatory metabolisms.
Carbon fixation in deep subsurface fluids
Although photosynthetically derived organic carbon can be found in Earth’s subsurface, it is often recalcitrant and a limiting nutrient for heterotrophs (Pedersen, 2000). Bioavailable, surface-derived organic carbon is likely limited at the deep sites in SURF, and hence, many resident heterotrophs must rely on in situ production of fixed carbon by chemolithoautotrophs, including nitrate reducers, methanogens, acetogens, sulfate reducers and iron reducers (Stevens and McKinley, 1995; Stevens, 1997; Pedersen, 2000; Lollar et al., 2006; Chivian et al., 2008; Beal et al., 2009; Magnabosco et al., 2015). As noted above, the most common mode of carbon fixation in the 74 MAGs was the reductive acetyl-CoA pathway. This ancient pathway is the only one known to be used by both Archaea and Bacteria (Hügler and Sievert, 2010). The predominance of this pathway was also documented in the metagenomic analysis of another terrestrial deep subsurface environment, the Witwatersrand Basin in South Africa (Magnabosco et al., 2015). That study concluded that the preference for the reductive acetyl-CoA pathway was in response to energy limitation, it being energetically inexpensive compared with the other five pathways (Berg, 2011; Hügler and Sievert, 2010) and hence ideal for organisms operating near the thermodynamic limit of life. Furthermore, the acetyl-CoA pathway requires anoxic conditions, as some of its enzymes, especially the crucial acetyl-CoA synthase, are highly oxygen sensitive (Berg, 2011). This pathway’s requirement for high levels of metals with low solubility under oxic or sulfidic conditions (Mo, Co, Ni, Fe) (Berg, 2011) also points to anoxic environments. Because of energetic efficiency and the necessity for anoxia, the acetyl-CoA pathway is the ideal mode of inorganic carbon fixation in highly reducing, aphotic and energy-deplete deep subsurface fluids, including those encountered at SURF, where the oxidation-reduction potential measured −235 to −276 mV (Osburn et al., 2014). Certainly, the relative dominance of the reductive acetyl-CoA pathway is, in part, because of the wide variety of organisms that have been reported to use this pathway, spanning both the Archaeal and Bacterial domains (Berg, 2011). Such organisms include acetogens, sulfate reducing bacteria, ammonia-oxidizing Planctomycetes and anaerobic facultative autotrophs (Schauder et al., 1988). Recently, the pathway was also shown to be run in reverse, with heterotrophs using carbon monoxide dehydrogenase and acetyl-CoA synthase to oxidize acetyl-CoA (Rabus et al., 2006), so it cannot be ruled out that some of the Bacteria found in SURF fluids are employing the reductive acetyl-CoA pathway heterotrophically.
Members of the phylum Chloroflexi commonly use the 3-hydroxypropionate bi-cycle for carbon fixation (Hügler and Sievert, 2010). In our seven Chloroflexi MAGs, however, evidence for this pathway was rare, limited to only one to two genes out of the 14 key genes in that cycle (Figure 3 and Supplementary Table 2). These results may be explained by the high energetic costs of this pathway; it requires seven ATP equivalents for the synthesis of pyruvate and three additional ATPs for the formation of triose phosphate (Berg, 2011). In many lineages of Chloroflexi, this high energy cost is offset by phototrophy, which is not possible in the dark subsurface at SURF. Instead, five Chloroflexi genomes contain the complete or near-complete reductive acetyl-CoA pathway (85–100% of genes) (Figure 3), which in contrast requires only 1 ATP and 2 NADPH reducing equivalents (Hügler and Sievert, 2010).
The canonical genes encoding enzymes requisite for the reduction of CO2 via the Calvin cycle, RuBisCo and phosphoribulokinase, were found in only one of our 74 MAGs (Figure 3). This MAG belongs to the phylum Proteobacteria. These translated genes were further investigated using BLASTp analysis and found to be closely related (93% identity over 99% query coverage) to the Type II RuBisCo found in typical Proteobacteria lineages (Hanson and Tabita, 2001; Tabita, et al., 2008). This would indicate that the Proteobacteria found in SURF fluids is likely capable of carbon fixation via the Calvin cycle and the cbbL annotation was not a false hit nor are these genes related to the Type IV Rubisco-like protein that has been shown not to fix carbon (Tabita et al., 2008).
Expansion of the predicted metabolic capabilities of the microbial dark matter and identification of two novel candidate phyla
In this study, we added four nearly complete (88–94%) MAGs to the repository of analyzed genomes in the candidate phylum Omnitrophica/OP3 (Rinke et al., 2013; Kolinko et al., 2015; Speth et al., 2016). This phylum was originally identified in a terrestrial hot spring, Obsidian Pool, in Yellowstone National Park, USA, leading to its designation ‘OP3’ (Hugenholtz et al., 1998). Since then, this phylum has been detected globally in environments such as flooded paddy soil (Derakshani et al., 2001), freshwater lakes and marine estuaries (Rinke et al., 2013), lake sediments (Kolinko et al., 2015), wastewater bioreactors (Speth et al., 2016), and the terrestrial subsurface (Rinke et al., 2013 and this study). Without cultured members, and with previously very little genetic sequence data to analyze, OP3 was placed within the Planctomycetes-Verrucomicrobia-Chlamydiaesuperphylum, along with Lentisphaerae (added later) (Wagner and Horn, 2006; Pilhofer et al., 2008). Our Omnitrophica and OP3 MAGs (SURF_12, 18, 26), include one of the most complete Ca. Omnitrophica genomes to date (93%, SURF_12). Similar to Rinke et al. (2013), we found genes for carbon fixation via the reductive acetyl-CoA pathway in all three MAGs.
In our 16 S rRNA and phylogenomic analyses, we observed incompatible polyphyly for MAGs SURF_12, 18, 25 and 26 with respect to the phyla OP3 and Omnitrophica. Previously, the phylum Omnitrophica was named on the basis of SAGs loosely related to OP3 16 S rRNA gene sequences from targeted gene surveys (Rinke et al., 2013). More recent studies have also grouped OP3 and Omnitrophica as a single phylum (Baker et al., 2015; Kolinko et al., 2015; Speth et al., 2016). However, after comparing all publically available OP3/Omnitrophica genomes, we conclude that OP3 and Omnitrophica are divergent. Based on extremely low (<30%) pairwise average amino-acid identity (data not shown) and phylogenomic analysis of concatenated single-copy genes (Figure 1), we propose that they be split into two separate phyla.
Based on our phylogenomic analysis using a concatenated alignment of single-copy marker genes (Figure 1), phylogenetic analysis of 16 S rRNA gene sequences (Supplementary Figure 2), and average amino-acid identity analyses, three MAGs did not fall within any previously described phylum. We propose that one MAG (SURF_26) represents the first genome of a novel phylum. With time, more related genomes from environmental datasets will likely become available, and we should then be able to better describe and name this phylum. The two other MAGs (SURF_5 and SURF_17) do not identify with any known phylum, either, and we propose that these belong to a newly described phylum within the domain Bacteria, here named SURF-CP-1.
The Zixibacteria (formerly RBG-1) were recently defined as a novel candidate phylum (Castelle et al., 2013). Sequences corresponding to this phylum have been identified in 16 S rRNA targeted surveys and metagenomic studies in global subsurface environments (Lin et al., 2012; Castelle et al., 2013; Baker et al., 2015). Most recently, numerous lineages within this phylum were found in the sulfate-methane transition zone in anoxic sediments of the eastern United States (Baker et al., 2015). Similar to previous studies (Castelle et al., 2013), we failed to identify a complete carbon fixation pathway in bin SURF_9, although the MAG is near (94%) complete. We consequently suggest that this candidate phylum heterotrophically scavenges reduced carbon for biomass synthesis (anabolism), and is capable of nitrate (narG), nitric oxide (norB) or sulfate (dsrAB) reduction, and possibly thiosulfate (sulfhydrogenase) disproportionation, as catabolic strategies (Figure 2).
Putative metabolisms in newly identified candidate phyla, SURF-CP-1 and -2
SURF-CP-1, the newly identified candidate phylum named Abyssubacteria, is composed of two genomes in this study, SURF_5 and SURF_17. These genomes are 96% and 91% complete, respectively. They contain homologs of cytoplasmic nitrate reductase (narABDG) (Figure 2). In addition, SURF_5 contains a putative nitric oxide reductase (norBC). We hypothesize that these Bacteria can use, nitrate or nitric oxide as electron acceptors. Interestingly, the two genomes have different profiles with respect to putative carbon fixation (Figure 3). SURF_5 contains 90% of the genes unique to the 3-hydroxypropionate/4-hydroxybutyrate bi-cycle and all genes necessary for the reductive Acetyl-CoA (Wood-Ljungdahl) pathway, whereas SURF_17 has only a complete reductive Acetyl-CoA pathway (Figure 3 and Supplementary Figure 1). This could be indicative of a highly versatile lifestyle, as the reductive Acetyl-CoA pathway can be utilized as both an autotrophic assimilatory metabolism and in reverse as a heterotrophic dissimilatory metabolism, as discussed above (Schauder et al., 1988; Rabus et al., 2006). SURF-CP-1 may even be using carbon monoxide as an electron donor for nitrate or sulfate reduction, redox couples that were predicted to be highly exergonic in these fluids by Osburn et al. (2014). SURF-CP-2 has a more cryptic lifestyle. The SURF_26 genome does not encode for any metabolic genes queried (Figure 2) nor does it have a complete carbon fixation pathway. It does not seem to be capable of a chemolithotrophic or autotrophic lifestyle, but could be heterotrophic or fermentative, metabolisms that were not as heavily investigated in this study.
Concluding remarks
This study used high-throughput Illumina sequencing to investigate a microbial ecosystem in the terrestrial DSB. Here, we find that Deltaproteobacteria and candidate phyla bacterial lineages are most abundant, with putative sulfate/sulfur reduction and nitrate/nitrite reduction likely being the most common energy metabolisms employed. This is consistent with previously calculated reaction energetics for deep subsurface fluids at SURF (Osburn et al., 2014). We also identified a surprisingly high relative abundance of candidate phyla in these deep subsurface fluids and identified two novel putative candidate phyla bacterial lineages (SURF-CP1 and SURF-CP-2). SURF-CP-1 has been given the name Abyssubacteria, the Latin prefix meaning ‘deep,’ as it was collected in the deep subsurface, and its closest relatives according to 16 S rRNA gene identity (98%) were found in the Nankai Trough and the world’s largest sink hole, located in central Mexico. SURF-CP-2 has been named Aureabacteria, Latin prefix meaning ‘gold’ in recognition that it was collected in the former Homestake gold mine.
Data deposit
Sequence data for metagenomic reads, contigs and genes were submitted to the JGI-IMG under accession numbers IMG 3300007354, 3300007352 and 3300007351 for SURF-B and –D fluids, and the combined assembly, respectively. Sample metadata can be accessed using the BioProject identifier PRJNA355136. The NCBI BioSamples used here are SAMN06064269 (SURF-B_fluid), SAMN06064270 (SURF-D_fluid), and SAMN06064271 (SURF_fluid_coasembly). FASTA files containing the contigs of all 74 MAGs can be accessed at doi: 10.6084/m9.figshare.4284578. A FASTA file containing 44 SSU rRNA genes with length >300 base pairs, including 40 extracted from the 74 MAGs, plus 4 additional SSU rRNA genes identified in preliminary (that is, non-reported) MAGs can be accessed at doi: 10.6084/m9.figshare.4284584. IMG/M-relevant files needed to isolate scaffold sets for all 74 genomes from metagenomes can be accessed at doi: 10.6084/m9.figshare.4284587.
References
Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ et al (2014). Binning metagenomic contigs by coverage and composition. Nat Methods 11: 1144–1146.
Aüllo T, Ranchou-Peyruse A, Ollivier B, Magot M (2013). Desulfotomaculum spp. and related gram-positive sulfate-reducing bacteria in deep subsurface environments. Front Microbiol 4: 362.
Baker BJ, Moser DP, MacGregor BJ, Fishbain S, Wagner M, Fry NK et al (2003). Related assemblages of sulphate‐reducing bacteria associated with ultradeep gold mines of South Africa and deep basalt aquifers of Washington State. Environ Microbiol 5: 267–277.
Baker BJ, Lazar CS, Teske AP, Dick GJ (2015). Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 3: 14.
Baker BJ, Saw JH, Lind AE, Lazar CS, Hinrichs K-U, Teske AP et al (2016). Genomic inference of the metabolism of cosmopolitan subsurface Archaea, Hadesarchaea. Nat Microbiol 1: 16002.
Beal EJ, House CJ, Orphan VJ (2009). Manganese- and iron-dependent marine methane oxidation. Science 325: 184–187.
Berg IA (2011). Ecological aspects of the distribution of different autotrophic CO2 fixation pathways. Appl Environ Microbiol 77: 1925–1936.
Bolger AM, Lohse M, Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120.
Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Schulz F, Doud D et al (2017). Genome standards for single amplified genomes and genomes from metagenomes of Bacteria and Archaea. Nat Biotechnol in press.
Campbell JH, O’Donoghue P, Campbell AG, Schwientek P, Sczyrba A, Woyke T et al (2013). UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc Natl Acad Sci USA 110: 5540–5545.
Castelle CJ, Hug LA, Wrighton KC, Thomas BC, Williams KH, Wu D et al (2013). Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat Commun 4: 2120.
Castelle CJ, Wrighton KC, Thomas BC, Hug LA, Brown CT, Wilkins MJ (2015). Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol 25: 690–701.
Chivian D, Brodie EL, Alm EJ, Culley DE (2008). Environmental genomics reveals a single- species ecosystem deep within Earth. Science 322: 275–278.
Cowen JP, Giovannoni SJ, Kenig F, Johnson HP, Butterfield D, Rappé MS et al (2003). Fluids from aging ocean crust that support microbial life. Science 299: 120–123.
Creevey CJ, Doerks T, Fitzpatrick DA, Raes J, Bork P (2011). Universally distributed single-copy genes indicate a constant rate of horizontal transfer. PLoS One 6: e22099.
Darling AE, Jospin G, Lowe E, Matsen FA IV, Bik HM, Eisen JA (2014). PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ 9: e243.
Derakshani M, Lukow T, Liesack W (2001). Novel bacterial lineages at the (sub)division level as detected by signature nucleotide-targeted recovery of 16 S rRNA genes from bulk soil and rice roots of flooded rice microcosms. Appl Environ Microbiol 67: 623–631.
Grimm F, Franz B, Dahl C (2008)Thiosulfate and sulfur oxidation in purple sulfur bacteriaIn Microbial Sulfur Metabolism. Springer: Berlin, Heidelberg, 101–116.
Dong Y, Kumar CG, Chia N, Kim PJ, Miller PA, Price ND et al (2014). Halomonas sulfidaeris‐ dominated microbial community inhabits a 1.8 km‐deep subsurface Cambrian Sandstone reservoir. Environ Microbiol 16: 1695–1708.
Dunham JP, Friesen ML (2013). A cost-effective method for high-throughput construction of Illumina sequencing libraries. Cold Spring Harbor Protocols 2013: 820–834.
Dupont C, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, Valas R et al (2012). Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6: 1186–1199.
Eddy SR (2011). Accelerated profile HMM searches. PLoS Comput Biol 10: e1002195.
Edwards RA, Rodriguez-Brito B, Wegley L, Haynes M, Breitbart M, Peterson DM et al (2006). Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 7: 57.
Eren AM, Esen ÖC, Quince C, Vineis JH, Morrison HG, Sogin ML et al (2015). Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3: e1319.
Fredrickson JK, Onstott TC (1996). Microbes deep inside the earth. Sci Am 275: 68–73.
Ghosh W, Dam B (2009). Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol Re 33: 999–1043.
Hanson TE, Tabita FR (2001). A ribulose-1, 5-bisphosphate carboxylase/oxygenase (RubisCO)-like protein from Chlorobium tepidum that is involved with sulfur metabolism and the response to oxidative stress. Proc Natl Acad Sci 8: 4397–4402.
Hugenholtz P, Pitulle C, Hershberger KL, Pace NR (1998). Novel division level bacterial diversity in a Yellowstone hot spring. Journal of Bacteriology 180: 366–376.
Hügler M, Sievert SM (2010). Beyond the Calvin cycle: autotrophic carbon fixation in the ocean. Marine Sci 3: 261–289.
Huntemann M, Ivanova NN, Mavromatis K, Tripp HJ, Paez-Espino D, Palaniappan K et al (2015). The standard operating procedure of the DOE-JGI Microbial Genome Annotation Pipeline (MGAP v. 4). Stand Genomic Sci 10: 86.
Hyatt D, LoCascio PF, Hauser LJ, Uberbacher EC (2012). Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28: 2223–2230.
Jungbluth SP, Grote J, Lin HT, Cowen JP, Rappé MS (2013). Microbial diversity within basement fluids of the sediment-buried Juan de Fuca Ridge flank. ISME J 7: 161–172.
Jungbluth SP, Glavina del Rio T, Tringe SG, Stepanauskas R, Rappé MS (2017). Genomic comparisons of a bacterial lineage that inhabits both marine and terrestrial deep subsurface systems. PeerJ 5: e3134.
Kallmeyer J, Pockalny R, Adhikari RR (2012). Global distribution of microbial abundance and biomass in subseafloor sediment. Proc Natl Acad Sci USA 109: 16213–16216.
Kantor RS, Wrighton KC, Handley KM, Sharon I, Hug LA, Castelle CJ et al (2013). Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. MBio 4: e708–e713.
Kolinko S, Richter M, Glöckner FO, Brachmann A, Schüler D (2015). Single‐cell genomics of uncultivated deep‐branching magnetotactic bacteria reveals a conserved set of magnetosome genes. Environ Microbiol 18: 21–37.
Langmead B, Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359.
Lau MCY, Cameron C, Magnabosco C, Brown CT, Schilkey F, Grim S et al (2014). Phylogeny and phylogeography of functional genes shared among seven terrestrial subsurface metagenomes reveal N-cycling and microbial evolutionary relationships. Front Microbiol 5: 531.
Lin LH, Wang PL, Rumble D, Lippmann-Pipke J, Boice E, Pratt LM et al (2006). Long-term sustainability of a high-energy, low-diversity crustal biome. Science 314: 479–482.
Lin X, Kennedy D, Fredrickson J, Bjornstad B, Konopka A (2012). Vertical stratification of subsurface microbial community composition across geological formations at the Hanford Site. Environ Microbiol 14: 414–425.
Lollar BS, Lacrampe-Couloume G, Slater GF, Ward J, Moser DP, Gihring TM et al (2006). Unravelling abiogenic and biogenic sources of methane in the Earth's deep subsurface. Chem Geol 22: 328–339.
Lovley DR, Chapelle FH (1995). Deep subsurface microbial processes. Rev Geophys 33: 365–381.
Loy A, Duller S, Baranyi C, Mußmann M, Ott J, Sharon I et al (2009). Reverse dissimilatory sulfite reductase as phylogenetic marker for a subgroup of sulfur‐oxidizing prokaryotes. Environ Microbiol 11: 289–299.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Buchner A et al (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
McMahon S, Parnell J (2014). Weighing the deep continental biosphere. FEMS Microbiol Ecol 87: 113–120.
Magnabosco C, Ryan K, Lau MC, Kuloyo O, Lollar BS, Kieft TL et al (2015). A metagenomic window into carbon metabolism at 3 km depth in Precambrian continental crust. ISME J 10: 730–741.
Matsen FA, Kodner RB, Armbrust EV (2010). pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11: 1–16.
Markowitz VM, Ivanova NN, Szeto E, Palaniappan K, Chu K, Dalevi D et al (2008). IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res 36: 534–538.
Markowitz VM, Chen IM, Chu K, Szeto E, Palaniappan K, Pillay M et al (2014). IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res 42: 568–573.
Momper LM, Reese BK, Carvalho G, Lee P, Webb EA (2015). A novel cohabitation between two diazotrophic cyanobacteria in the oligotrophic ocean. ISME J 4: 882–893.
Nyyssönen M, Hultman J, Ahonen L, Kukkonen I, Paulin L, Laine P et al (2014). Taxonomically and functionally diverse microbial communities in deep crystalline rocks of the Fennoscandian shield. ISME J 8: 126–138.
Onstott TC, Phelps TJ, Colwell FS, Ringelberg D, White DC, Boone DR et al (1998). Observations pertaining to the origin and ecology of microorganisms recovered from the deep subsurface of Taylorsville Basin, Virginia. Geomicrobiol J 15: 353–585.
Orcutt BN, Sylvan JB, Knab NJ, Edwards KJ (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol Mol Biol Rev 75: 361–422.
Osburn MR, LaRowe DE, Momper L, Amend JP (2014). Chemolithotrophy in the continental deep subsurface: Sanford Underground Research Facility (SURF), USA. Front Extr Microbiol 5: 610.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Gen Res 25: 1043–1055.
Peng Y, Leung HC, Yiu SM, Chin FY (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28: 1420–1428.
Pedersen K (2000). Exploration of deep intraterrestrial microbial life: current perspectives. FEMS Microbiol Lett 185: 9–16.
Pfiffner SM, Cantu JM, Smithgall A, Peacock AD, White DC, Moser DP et al (2006). Deep subsurface microbial biomass and community structure in Witwatersrand Basin mines. Geomicrobiol J 23: 431–442.
Pilhofer M, Rappl K, Eckl C, Bauer AP, Ludwig W, Schleifer KH et al (2008). Characterization and evolution of cell division and cell wall synthesis genes in the bacterial phyla Verrucomicrobia, Lentisphaerae, Chlamydiae, and Planctomycetes and phylogenetic comparison with rRNA genes. J Bacteriol 190: 3192–3202.
Price MN, Dehal PS, Arkin AP (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5: e9490.
Pruesse E, Queast C, Knittel K, Fuchs BM, Ludwig W, Peplies J et al (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196.
Pruesse E, Peplies J, Glöckner FO (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28: 1823–1829.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P et al (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41: 590–596.
Rabus R, Hansen TA, Widdel F (2006). Dissimilatory sulfate- and sulfur-reducing prokaryotes. The Prokaryotes Springer: New York, NY, USA, pp 659–768.
Reed AJ, Lutz RA, Vetriani C (2006). Vertical distribution and diversity of bacteria and archaea in sulfide and methane-rich cold seep sediments located at the base of the Florida Escarpment. Extremophiles 10: 199–211.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF et al (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437.
Sander J, Engels-Schwarzlose S, Dahl C (2006). Importance of the DsrMKJOP complex for sulfur oxidation in Allochromatium vinosum and phylogenetic analysis of related complexes in other prokaryotes. Arch Microbiol 186: 357–366.
Schauder R, Preuß A, Jetten M, Fuchs G (1988). Oxidative and reductive acetyl CoA/carbon monoxide dehydrogenase pathway in Desulfobacterium autotrophicum. Arch Microbiol 151: 84–89.
Seitz KW, Lazar CS, Hinrichs K-U, Teske AP, Baker BJ (2016). Genomic reconstruction of a novel, deeply branched sediment archaeal phylum with pathways for acetogenesis and sulfur reduction. ISME J 10: 1696–1705.
Simkus DN, Slater GF, Lollar BS, Wilkie K, Kieft TL, Magnabosco C et al (2016). Variations in microbial carbon sources and cycling in the deep continental subsurface. Geochimica et Cosmochimica Acta 173: 264–283.
Speth DR, Guerrero-Cruz S, Dutilh BE, Jetten MS (2016). Genome-based microbial ecology of anammox granules in a full-scale wastewater treatment system. Nature Communications 7: 1–10.
Stamatakis A (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690.
Stevens T (1997). Lithoautotrophy in the subsurface. FEMS Microbiol Rev 20: 327–337.
Stevens TO, McKinley JP (1995). Lithoautotrophic microbial ecosystems in deep basalt aquifers. Science 270: 450–454.
Tabita FR, Satagopan S, Hanson TE, Kreel NE, Scott SS (2008). Distinct form I, II, III, and IV RuBisCo proteins from the three kingdoms of life provide clues about RuBisCo evolution and structure/function relationships. J Exp Botany 59: 1515–1524.
Tiago I, Veríssimo A (2013). Microbial and functional diversity of a subterrestrial high pH groundwater associated to serpentinization. Environ Microbiol 15: 1687–1706.
Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM et al (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428: 37–43.
Wagner M, Horn M (2006). The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr Opin Biotechnol 17: 241–249.
Whitman WB, Coleman DC, Wiebe WJ (1998). Prokaryotes: the unseen majority. Proc Natl Acad Sci USA 95: 6578–6583.
Wu M, Scott AJ (2012). Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 28: 1033–1034.
Yarza P, Richter M, Peplies J, Euzeby J, Amann R, Schleifer KH et al (2008). The All-Species Living Tree project: A 16 S rRNA-based phylogenetic tree of all sequenced type strains. Syst Appl Microbiol 31: 241–250.
Youssef NH, Rinke C, Stepanauskas R, Farag I, Woyke T, Elshahed MS (2015a). Insights into the metabolism, lifestyle and putative evolutionary history of the novel archaeal phylum 'Diapherotrites'. ISME J 9: 447–460.
Acknowledgements
This work was supported by the NASA Astrobiology Institute under cooperative agreement NNA13AA92A. Many thanks to John Heidelberg and Rohan Sachdeva at USC for use of their servers and help in metagenomic analysis. We would also like to recognize A Murat Eren for his invaluable help in utilizing the Anvi’o interface. We want to thank especially staff and personnel at SURF for access to the deep subsurface and repeated access to samples used in this study. This work was funded by the NASA Astrobiology Institute under cooperative agreement NNA13AA92A.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Momper, L., Jungbluth, S., Lee, M. et al. Energy and carbon metabolisms in a deep terrestrial subsurface fluid microbial community. ISME J 11, 2319–2333 (2017). https://doi.org/10.1038/ismej.2017.94
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.94
This article is cited by
-
Deep subsurface igneous rocks from the Deccan traps harbor H2 and CO2 utilizing chemolithoautotrophic bacteria
Biologia (2024)
-
Unveiling the dynamics of the breast milk microbiome: impact of lactation stage and gestational age
Journal of Translational Medicine (2023)
-
Chemolithoautotroph distributions across the subsurface of a convergent margin
The ISME Journal (2023)
-
Hyperactive nanobacteria with host-dependent traits pervade Omnitrophota
Nature Microbiology (2023)
-
Subsurface hydrocarbon degradation strategies in low- and high-sulfate coal seam communities identified with activity-based metagenomics
npj Biofilms and Microbiomes (2022)
