
Abstract
All animals live in intimate association with communities of microbes, collectively referred to as their microbiota. Certain host traits can influence which microbial taxa comprise the microbiota. One potentially important trait in vertebrate animals is the adaptive immune system, which has been hypothesized to act as an ecological filter, promoting the presence of some microbial taxa over others. Here we surveyed the intestinal microbiota of 68 wild-type zebrafish, with functional adaptive immunity, and 61 rag1− zebrafish, lacking functional B- and T-cell receptors, to test the role of adaptive immunity as an ecological filter on the intestinal microbiota. In addition, we tested the robustness of adaptive immunity’s filtering effects to host–host interaction by comparing the microbiota of fish populations segregated by genotype to those containing both genotypes. The presence of adaptive immunity individualized the gut microbiota and decreased the contributions of neutral processes to gut microbiota assembly. Although mixing genotypes led to increased phylogenetic diversity in each, there was no significant effect of adaptive immunity on gut microbiota composition in either housing condition. Interestingly, the most robust effect on microbiota composition was co-housing within a tank. In all, these results suggest that adaptive immunity has a role as an ecological filter of the zebrafish gut microbiota, but it can be overwhelmed by other factors, including transmission of microbes among hosts.
Similar content being viewed by others

Introduction
Adaptive immunity is a fascinating biological phenomenon that exists only in jawed vertebrates. Its defining characteristics are its ability to modify its receptor repertoire during the course of an individual’s life, and to ‘remember’ encounters with previous pathogens, making it much easier to prevent or clear subsequent infections by the same pathogen (Flajnik and Kasahara, 2010). The role of adaptive immunity in disease resistance is well documented; however, its role in shaping commensal microbial communities is not well understood. An emerging conceptual model for this role is that of an ‘ecological filter’. The concept of an ecological filter is common to plant ecology and can be defined in that context as ‘a sieve that filters out individuals from an initial seed population during successive life stages and ultimately determines the success of a seed in producing more seeds’ (George and Bazzaz, 1999). In the context of host–microbe associations, we can view the host as the environment imposing ecological filters, and the microbial communities as the seeds undergoing this filtering. Given the benefits provided to the host by certain bacterial taxa and the desirability of selecting against pathogenic microorganisms, it is in the host’s interest to filter which microbial taxa comprise its microbiota. Many host factors could act as ecological filters on the microbiota, from literal physical barriers such as intestinal mucus (Johansson et al., 2008), to more abstract filters such as diet (Turnbaugh et al., 2008; Wong et al., 2015). These host factors may be redundant, or may interact in either additive or non-additive ways. Here we explore the specific contribution of the adaptive immune system to filtering the intestinal microbial community in zebrafish.
There is a growing body of evidence that the immune system can act as an ecological filter of the microbiota in humans and animal models. Genome-wide association studies and quantitative trait loci mapping studies have found significant correlations between immune genes and the composition of the human and mouse microbiota (Benson et al., 2010; Spor et al., 2011; Kostic et al., 2013; Davenport et al., 2015; Org et al., 2015; Goodrich et al., 2016). Furthermore, it has been demonstrated that immune receptors physically interact with the gut microbiota, influencing its composition and diversity in ways that affect the health of the host (Palm et al., 2014; Bunker et al., 2015; Fransen et al., 2015; Kubinak et al., 2015a,2015b). Direct manipulations of the host adaptive immune system have also revealed significant changes in the gut microbiota (Dimitriu et al., 2013; Brugman et al., 2014; Kawamoto et al., 2014; Shen et al., 2014; Zhang et al., 2015). However, such studies, particularly those in mice, often utilize small sample sizes, or involve designs that cannot distinguish genotype-specific effects from those associated with isolation of genotypes in distinct cages, and few studies have looked explicitly at the strength of ecological filtering among cage mates (Dimitriu et al., 2013; Fransen et al., 2015). Similar to this current study, Dimitriu et al. (2013) compared the gut microbiota between wild-type and immunocompromised hosts that were either separated by genotype or mixed. They found that when genotypes were mixed, wild-type gut communities became more like immunocompromised gut communities, implying that the filtering effects of adaptive immunity can be overwhelmed by migration from other host communities. However, without replicating treatments, the possibility of differences in gut bacterial communities arising in part from cage effects (that is, random or deterministic effects on the bacterial communities due to physically separated housing units) is still present in such a study. Human studies that show differences in host-associated communities of the skin (Oh et al., 2013) or gut of immunocompromised patients (Daniels et al., 2007; Gori et al., 2008), utilize larger sample sizes than many mouse studies, but lack control over environmental conditions and genetic backgrounds of subjects.
The zebrafish is an excellent model for testing the filtering effect of adaptive immunity because many of the shortcomings of mouse and human studies can be simultaneously addressed. The zebrafish possesses an adaptive immune system very similar to that of mammals (Rauta et al., 2012), a large number of individuals from a single breeding pair can be used in a single experiment, housing conditions can be readily manipulated, and the shared environment can be exhaustively sampled. Although innate immunity is active at the time of hatching, adaptive immunity in zebrafish does not become fully functional until between 21 and 28 d.p.f. (days post fertilization; Lam et al., 2004), allowing experimenters to more easily disentangle the unique contributions of each type of immunity. Recently, we documented how the composition of the gut microbiota changes throughout zebrafish development (Stephens et al., 2016). There were significant changes in microbiota composition after 21 d.p.f., despite constant housing conditions and diet, which implicates adaptive immunity as a possible ecological filter shaping the gut microbiota. In addition, we inferred the relative roles of neutral and selective processes in shaping the zebrafish gut microbiota over development, by fitting the data to an ecological model that assumes only neutral community assembly processes (such as dispersal and stochastic loss of individuals). We previously reported a trend of decreasing neutral model fit across development, implying that ecological filters become more important in shaping the gut microbiota as the zebrafish develops (Burns et al., 2016). From these results, we suspect that one of the ways that hosts increase filtering of their gut microbiota through time is via the maturation of the adaptive immune system.
To determine whether adaptive immunity is an important ecological filter of the gut microbiota in the adult zebrafish, we compared the gut bacterial communities of wild-type (adaptive immunity present) and rag1− (adaptive immunity inactive) hosts (Wienholds, 2002). We had four main hypotheses regarding adaptive immunity’s role as a filter. (1) Adaptive immunity, due to the somatic recombination of B- and T-cell receptors, has an individualizing effect on the gut microbiota of each host, resulting in greater variation in community composition among wild-type hosts than rag1− hosts. (2) The filtering effects of adaptive immunity will lead to distinct differences between the composition of wild-type and immune-deficient hosts. (3) Adaptive immunity acts as a filter of microorganisms in the host’s environment, resulting in a greater difference between environmental water communities and the intestinal microbiota of hosts with a functional immune system versus those without. (4) The lack of filtering by adaptive immunity in rag1− hosts results in a greater role for neutral assembly processes in the gut microbiota.
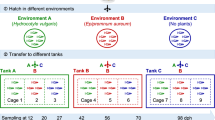
Because of the potential for transmission of microorganisms and/or host factors among hosts in a shared environment, we suspected that the ability of the adaptive immune system to act as an ecological filter could be altered by the presence of other fish. We therefore created a treatment with either a low or high potential for transmission between wild-type and rag1− hosts by segregating or mixing genotypes, respectively (Figure 1). We hypothesized that increasing the potential for transmission between wild-type and rag1− hosts by co-housing genotypes would overwhelm the effects of adaptive immunity, making their communities more similar to rag1− communities, as demonstrated in mice by Dimitriu et al. (2013). To measure stochastic tank-specific effects (differences due to variability between tanks themselves), we maintained three tanks of each housing by genotype treatment.

Experimental design for comparison of wild-type and rag1− gut microbiota. To avoid generating 50% heterozygous offspring we cross two set of parents: two rag1+/+ parents and two rag1−/− parents. These sets of parents were generated from the same line, maintained as heterozygotes. We housed each genotype separately and together, and each housing treatment was replicated three times.
Materials and methods
Experimental design and sample collection
We crossed two pairs of fish derived from the same line (Wienholds, 2002); one pair were both rag1+ (wild type) and the other pair were both rag1−. We previously confirmed the lack of mature immunoglobulin production in the rag1− mutants via PCR. Using primers specific to various immunoglobulin V segments of the variable region and reverse primers specific to the immunoglobulin M constant regions, we attempted to amplify full-length immunoglobulin M transcripts in wild- type and rag1− samples (one primer pair per reaction). Wild-type samples all produced transcripts of the expected sizes while rag1− samples produced no amplicons (primer sequences in Supplementary Table S1). We raised nine tanks of 22 fish each, on standard continuous flow in the UO aquatic animal facility: three tanks of only wild-type, three tanks of only rag1− and three tanks with equal numbers of both genotypes. Although tank water was recirculated through the facility, it first passed through a sand and ultraviolet filter designed to limit the transmission of microorganisms among tanks. At 9 d.p.f., six fish from each tank were removed for a separate experiment. At 75 d.p.f., approximately a month after adaptive immunity should have become fully functional in all wild-type fish, all the remaining samples (134) were killed for gut dissection and microbial DNA extraction. After the fish were removed from the tank, the entire water volume of each tank was run through a filter. Each filter was then subjected to DNA extraction. Water and zebrafish sample handling during collection, dissection and DNA extraction were all performed as previously described (Stephens et al., 2016).
Carcasses of all fish were also kept after dissection, stored in TRIzol (Life Technologies, Carlsbad, CA, USA) at −80 °C. A subset of the segregated wild-type and rag1− samples were genotyped, via PCR (Wienholds, 2002), to confirm their presumed identity, and all mixed-housing samples were genotyped to determine their identity. Five mixed-housing samples with ambiguous genotyping results were excluded from analysis after sequencing. These five zebrafish appeared to be heterozygotes, but this could simply be a result of the genotyping protocol. One of the primers used in the PCR protocol creates a KpnI site in the amplicons from the wild-type allele but not the mutant allele. These non-complementary bases can sometimes be edited by the polymerase, thus making wild-type samples appear heterozygous. Given that we found no heterozygotes in the rag1− only housing, we deemed it unlikely that heterozygotes had contaminated our experiment but removed these apparent heterozygotes from the analysis as a precaution.
Illumina library preparation and 16S rRNA gene sequence analysis
We characterized the microbial communities of individual samples via Illumina (San Diego, CA, USA) sequencing of 16S rRNA gene amplicons. To prepare amplicons for Illumina sequencing, we used a single-step PCR method to add dual indices and adapter sequences to the V4 region of the bacterial 16S rRNA gene and generate paired-end 250 nucleotide reads on the Illumina HiSeq 2000 platform. Illumina sequence reads have been deposited under the NCBI SRA accession number SRP096739.
The 16S rRNA gene Illumina reads were processed using methods implemented by FLASH (Magoc and Salzberg, 2011), the FASTX Toolkit (Hannon Lab, 2010), and the USEARCH pipeline (Edgar, 2010). The processing pipeline can be found at http://github.com/kstagaman/Process_16S. Operational taxonomic units were defined using 97% sequence similarity. Read assembly, quality control and operational taxonomic unit table building were done on the University of Oregon ACISS cluster, and all subsequent data processing and diversity analysis were done in R (R Core Team, 2015).
Diversity measures and statistical tests
Host and water samples were processed together. Samples with fewer than 2000 total reads were removed from analysis (the mean read number for gut samples was 201 020, for water samples the mean was 106 049), and operational taxonomic unit abundances of the remaining samples were variance-stabilized using phyloseq (McMurdie and Holmes, 2013) and DESeq2 (Love et al., 2014) as recommended by McMurdie and Holmes (2014). Phylogenetic diversity was measured using Faith’s PD (Faith, 1992) as implemented in the picante package (Kembel et al., 2010). Weighted Unifrac distances (Lozupone and Knight, 2005) between communities were calculated and distance-based redundancy analysis (db-RDA) ordinations performed using the capscale function from the vegan package (Oksanen et al., 2016). Other distance metrics were used and produced very similar results. Permutational multivariate analysis of variance (ANOVA) tests on weighted Unifrac distances were performed using the adonis function from the vegan package. For non-distance data, analysis of variance and post hoc tests of significance were performed using the aov and TukeyHSD functions, respectively, from the R base packages when the number of observations in each treatment was similar. When sample sizes were not similar, analysis of variance with type III sum of squares was performed using the Anova function from the car package (Fox and Weisberg, 2011), and post hoc tests were performed using the DTK.test (Dunnett–Tukey–Kramer, DTK) function from the DTK package (Lau, 2013). Diversity data visualization was done with the ggplot2 package (Wickham, 2009).
To better understand the ecological processes involved in ecological filtering via the host adaptive immune system, we fit our microbiota data to an ecological model that assumes only neutral community assembly processes (such as dispersal and stochastic gain and loss of individuals). In brief, we fit the Sloan Neutral Community Model for Prokaryotes to the distribution of bacterial taxa in our data and assessed the goodness of fit (Sloan et al., 2006). The model predicts that taxa with a high abundance in a source pool of potential bacterial colonists will be found in a greater fraction of those hosts because, being more abundant, they are more likely to disperse by chance, while taxa of low average abundance are more likely to be lost from individual hosts due to stochastic processes and a lower likelihood of random dispersal. As we were primarily interested in determining how adaptive immunity mediates the filtering of the microbiota relative to other hosts in the population lacking adaptive immunity, rather than between hosts and their environment, we defined the source pool by the average abundance of bacterial taxa across all hosts of both genotypes and housing treatments and did not include the external environment. We inferred poor model fits to be indicative of non-neutral processes, such as host filtering, being important in distinguishing genotypes and housing treatments from one another. Because model fit can be influenced by the number of samples in each treatment, we ran the model on multiple subsets of samples of each treatment. Taxa whose frequency of occurrence across fish were inconsistent with the neutral model, falling outside the 95% confidence interval of the model prediction, were inferred to be subject to host filtering. For example, taxa that are ‘under represented’, that is present in fewer hosts than expected from their mean abundance, could be potential pathogens: successfully prevented from colonizing most hosts but achieving high abundance once established. Taxa that are ‘over represented’, that is found in more hosts than expected from their mean abundance, could be beneficial taxa actively promoted by the host. Using these cutoffs from the model, we partitioned communities into taxa with neutral and non-neutral distributions and determined whether the composition of these partitions differed by host genotype and housing conditions.
Results
Adaptive immunity acts as an individualizing filter
One of the hallmarks of adaptive immunity is the somatic rearrangement and hypermutation of T- and B-cell receptors. These processes allow even genetically identical hosts to possess quite different adaptive immune receptor repertoires (Weinstein et al., 2009). Different adaptive repertoires could theoretically filter the gut microbiota in different ways within each wild-type host, while other host factors (in a near-isogenic population) should filter the gut microbiota similarly across all hosts. We therefore hypothesized that adaptive immunity acts as an individualizing filter, making wild-type hosts more dissimilar, on average, from other wild-type hosts than rag1− hosts would be from each other. When we compared the taxonomic similarity of the microbiota (beta-diversity) between hosts of the same genotype across both housing treatments, we indeed found that wild-type hosts had a slightly, but significantly, greater average dissimilarity than rag1− hosts (FHost Genotype=14.41, P<0.001; Figure 2). This difference in similarity between genotypes maintained significance for hosts in the segregated housing treatment (DTK test P<0.05). For the mixed genotypes, we expected two possible outcomes. If adaptive immunity were a robust filter, we expected to see no change in its individualizing effect on the wild type and/or an overall decrease in the similarity of wild-type microbiota as they would be exposed to a more diverse pool from which to individually sample from. Alternatively, if it were not robust, we expected dispersal from rag1− hosts to make the wild-type hosts more homogenous. If dispersal between hosts were minimal, we expected no change in the homogeneity of either host genotype. However, contrary to either of those expectations, both wild-type hosts and rag1− hosts were significantly less homogenous when the genotypes were mixed compared with when each genotype was segregated (Figure 2; FHousing=51.86, P<<0.001). Furthermore, the difference in homogeneity between genotypes was not significantly different when they were in mixed housing (DTK test, P>0.05).

Pairwise weighted Unifrac distances between gut communities. We compared the average pairwise weighted Unifrac distance between gut communities within experimental treatments. Shared significance letters indicate no significant difference in means according to the DTK post hoc test.
Adaptive immunity does not significantly alter composition of the gut microbiota
The absence of some or all adaptive immune function has been associated with changes in gut microbiota alpha-diversity (number and abundance of different taxa within a community) in some mouse studies, although there are conflicting reports as to the direction of this change (Kawamoto et al., 2014 Supplementary Material; Shen et al., 2014; Zhang et al., 2015), and other studies report no or minimal effects of adaptive immunity on alpha-diversity (Dimitriu et al., 2013; Thoene-Reineke et al., 2014). We tested the hypothesis that adaptive immunity in zebrafish alters the alpha-diversity of the gut microbiota. We found no statistically significant differences between genotypes in either housing condition with regard to alpha-diversity (Figure 3a). We did find, however, that rag1− and wild-type hosts in mixed housing together had increased phylogenetic diversity (FHousing=10.18, P<0.01; Figure 3a), an outcome to be expected if two dissimilar communities become connected by dispersal (Mouquet and Loreau, 2002). We also measured the phylogenetic diversity of each tank environment (water), and while there was no significant difference in the mean diversity (FTank Genotype=1.68, P=0.26), this lack of significance is likely due to only having three samples per treatment (Figure 3b). There is a significant difference in variance of phylogenetic diversity between the water samples (Bartlett Test K2=8.94, P=0.01), with the mixed-housing treatment appearing to be the most variable. Similarly, when the distance between water samples is compared within each treatment, the mixed tanks trend toward the greatest dissimilarity, and the rag1−-only tanks trend toward the most homogeneous, although this result is also not statistically significant (possibly due to a lack of statistical power; Supplementary Figure S1), again congruent with the expectation of increased phylogenetic diversity due to the merging of two dissimilar communities. Nevertheless, permutational multivariate ANOVA analysis of community distance metrics was unable to distinguish gut bacterial communities by genotype, housing condition or their interaction (Table 1, Supplementary Figure S2). In fact, the only significant factor, which explained roughly 9% of the variance in gut communities across all samples, was the specific tank (labeled A through I) in which the zebrafish were housed (FTank=1.33, P<0.05; Table 1,Supplementary Figure S2). Consistent with the permutational multivariate ANOVA results, analysis of the ‘core’ microbiota (genera present in ⩾80% of samples) revealed similar core taxa for each genotype-by-housing treatment (Table 2). These taxa were also largely consistent with the most abundant genera found in Roeselers et al. (2011). Of particular note, Mycobacterium, a common fish pathogen, was a core genus for all treatments except the segregated wild-type hosts, suggesting a classical role for adaptive immunity in protecting against pathogens in zebrafish at both the individual and population level.

Phylogenetic alpha-diversity of gut and water communities. We compared the average phylogenetic diversity (PD) of each experimental treatment for (a) gut samples and (b) water samples. The double asterisk indicates a significant difference with P<0.01 according to ANOVA. NS, not significant.
Adaptive immunity does not significantly filter environmental bacteria
We hypothesized that adaptive immunity is an important ecological filter on the gut microbiota, with the result that the gut microbiota of wild-type hosts differ more from the environmental bacterial community than do the gut microbiota of rag1− hosts. To test this hypothesis, we compared the average beta-diversity, that is, differences in the abundance and number of taxa between communities, between the gut microbiota of individual hosts and the bacterial communities of their tank water. As shown in Supplementary Figure S3, we found no significant difference in the similarity between gut and water bacterial communities among the wild-type and rag1− hosts. We did see an effect of housing treatment (segregated versus mixed genotypes) on the similarity between gut and water communities; however, neither this difference nor the direction of its effect was robust to the distance metric used (Supplementary Figure S3), making the interpretation of these results difficult.
Adaptive immunity increases the role of selective processes in assembly of the gut microbiota
To further investigate the role of adaptive immunity, we used a neutral model to ask whether the presence of adaptive immunity increases the degree of filtering among hosts of each genotype. For each genotype, we asked whether a neutral model could predict the distribution of microbial taxa across hosts by the average abundance of those taxa across hosts of all genotypes and housing treatments (see Supplementary Figure S4A for an example of the analysis). We previously documented a decrease in the fit of a neutral model for distributions of zebrafish gut microbiota members through developmental time, including time after the onset of adaptive immunity (Burns et al., 2016). We therefore hypothesized that the presence of adaptive immunity, acting as an ecological filter, is one of the host factors contributing to the reduced model fit to the distributions of gut microbial taxa. A lower fit of the model for the wild-type compared with rag1− hosts in our study would indicate that adaptive immunity increases the strength of filtering between hosts and the source pool of all potential fish-associated microbes and thus support our hypothesis.
Our analysis revealed that, whether genotypes were segregated or mixed, the fit of the neutral model was lower for wild-type hosts than rag1− hosts (Figure 4). In addition, the model fit for both mixed genotypes appears to increase slightly from the model fits for both segregated genotypes.

R2 values for fit of neutral model to each tank. Boxplots represent quantiles for goodness of fit of 99 random bootstraps. Bootstrapping is necessary because fit can be influenced by the total number of samples in a treatment, and thus each treatment must be subsetted to an equal number of samples. Because the number of bootstraps can influence statistical inference, no statistics were applied to the goodness of fit.
We further investigated the neutral model to determine whether there were differences in the subsets of taxa whose distributions deviated from the expectation of the neutral model. The neutral model analysis can be used to assign each taxon into a ‘neutral’ or ‘non-neutral’ partition based on its distribution (Supplementary Figure S4A). The ‘non-neutral’ partition can be further subdivided into ‘over represented’ if the taxon is found in more hosts than expected based on its abundance and ‘under represented’ if the taxon is found in fewer hosts than expected based on its abundance. We divided the gut microbiota of each genotype-by-housing treatment into ‘over represented’, ‘neutral’ and ‘under represented’ partitions and compared the lists of taxa found in each partition of each treatment using the Sørensen distance (a presence–absence based distance metric suited to such a task; Sørensen, 1948). To confirm that differences between genotypes and housing treatments were not simply due to the effect of partitioning itself, we compared each observed distance to the mean distance of a series of simulated partition sampling as described in the the caption for Supplementary Figure S4. Supplementary Figure S4B illustrates the Sørensen distances between partitioned communities and reveals that the housing treatment effects the distance between the two ‘non-neutral’ partitions of each genotype, but not the ‘neutral’ partition, with the largest distance between genotypes in the ‘over represented’ partition within segregated housing and the smallest distance is in the ‘under represented’ partition, also within segregated housing.
Discussion
The role that adaptive immunity has in host–pathogen interaction has been well established, but its role in shaping the commensal microbiota is less understood. Although there is growing evidence that adaptive immunity can act as an ecological filter of the commensal gut microbiota, the nature and relative strength of this filtering is not known. We endeavored to rigorously examine adaptive immunity’s filtering effects utilizing the zebrafish gut microbiota as a model. We observed that adaptive immunity individualizes the gut microbiota of hosts, that is, hosts lacking adaptive immunity become more similar to each other than wild-type hosts. This effect of genotype maintains significance within the segregated housing treatment, but not the mixed-housing treatment, which is consistent with a possible mechanism wherein dispersal from wild-type hosts causes rag1− hosts to become more individualized.
There was no significant difference in phylogenetic diversity between genotypes either across or within each housing treatment, but both genotypes exhibited greater diversity in mixed housing over segregated housing. In a similar vein, the phylogenetic diversity of the water samples in the mixed-housing treatment trend higher than the segregated housing treatment water samples, although this difference is not statistically significant. Both these results are consistent with the prediction that if two dissimilar communities become connected by dispersal, both communities will increase in diversity due to the introduction of new taxa into each (Mouquet and Loreau, 2002). In this case, the water may exhibit an increase in diversity as a consequence of being the medium for the microbial dispersal between hosts. We also compared the community dissimilarity between the gut samples and the water samples, but found no effect of genotype. We did find effects of housing treatment, but the direction of this effect was sensitive to the distance metric used, rendering interpretation of this result difficult.
The increase in phylogenetic diversity in the mixed-housing treatment led us to hypothesize that differences in community composition between genotypes would be detectable by permutational multivariate ANOVA analysis of the pairwise ecological distances. However, our analyses revealed no statistically discernable difference in the overall community composition between the two genotypes. Rather, we found the variable that explained the greatest amount of variation in community composition was the specific tank in which the hosts were housed. This result highlights the importance of replication across housing units (tanks or cages) in future work, as differences in gut microbiota may be attributed to differing host genotypes when they might rather simply be due to housing effects. This lack of difference in community composition was particularly unexpected given the seemingly strong effects of adaptive immunity on the gut microbiota in previous studies (Dimitriu et al., 2013; Brugman et al., 2014; Kawamoto et al., 2014; Shen et al., 2014; Zhang et al., 2015). One possible explanation for this discrepancy is that the zebrafish adaptive immune system differs in certain features from the mammalian immune system. The zebrafish adaptive immune system develops only three immunoglobulin classes, does not undergo class switching and has less efficient affinity maturation (Traver et al., 2003). In addition, there is evidence that natural killer cells, a part of the innate immune system, are able to provide specific protection after bacterial re-exposure, which may at least partially compensate for the lack of functional B and T cells in rag1− hosts (Hohn and Petrie-Hanson, 2012). Brugman et al. (2014), did find, however, a significant effect of the presence of adaptive immunity on the zebrafish gut microbiota, although their sample size was much smaller and lacked replication.
Our most striking finding of a difference between wild-type and rag1− fish, that the rag1− hosts have greater inter-subject similarity, could be explained by two possible mechanisms. The rag1− hosts could be more homogeneous because, lacking an adaptive immune system, the innate immune system compensates and applies a greater selective pressure, that is similar across hosts, on the gut microbiota. In contrast, the rag1− hosts could be more homogenous because, lacking the adaptive immune system filter, they more neutrally sample the shared bacterial community from their external environment, resulting in similar distributions of taxa. To test these two hypotheses, we applied a neutral model to the distribution of bacterial taxa in our data and assessed its fit. Our finding that the neutral model has a greater fit to rag1− gut community distributions in both housing treatments suggests the latter hypothesis: the absence of the adaptive immune system removes the ability of hosts to filter their gut microbiota. It should be noted that while neutral processes have a more important role in microbiota assembly in rag1− hosts than wild-type hosts, this does not mean that other selective processes are not important, as the model typically explains less than 40% of the variance seen in the taxon distributions.
The adaptive immune system has a more subtle and complicated effect on the composition of the zebrafish gut microbiota than initially expected. In particular, the main effect of the adaptive immune system appears to be to individualize the microbiota without shifting their average composition as a whole, as evident by the observed differences between genotypes in the similarity among individuals, but not in their overall composition or phylogenetic diversity. Reasons for this departure from expectation might be the greater number of individuals and treatment replications included in this study relative to previous ones. Previous work that demonstrated a strong effect of adaptive immunity may have been due to conflation of tank and genotype effects, which our study design avoids. Furthermore, the relatively subtle effect we observed would likely not have been detected without the large number of individuals and high degree of replication possible in our study relative to previous studies of the effect of adaptive immunity on microbiomes.
The role of the adaptive immune system as an ecological filter is further supported by the increased fit of a neutral assembly model to the gut microbiota of rag1− mutant hosts. However, these effects are complicated by the overwhelming influence of the environment and exposure to different hosts, which had much stronger effects on phylogenetic diversity, community composition and inter-individual similarity. This suggests adaptive immunity does have a role in selecting the gut microbiota of zebrafish, but differences in the environment, in this case the tank, can obfuscate its effects. Our study highlights the importance of considering the interactions among genotype, environment and microbial dispersal when designing experiments for studying host–microbe systems. Future investigations on the interaction between adaptive immunity and the microbiota will need to develop more sophisticated frameworks to overcome these challenges.
References
Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J et al. (2010). Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA 107: 18933–18938.
Brugman S, Schneeberger K, Witte M, Klein MR, van den Bogert B, Boekhorst J et al. (2014). T lymphocytes control microbial composition by regulating the abundance of Vibrio in the zebrafish gut. Gut Microbes 5: 737–747.
Bunker JJ, Flynn TM, Koval JC, Shaw DG, Meisel M, McDonald BD et al. (2015). Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity 43: 541–553.
Burns AR, Stephens WZ, Stagaman K, Wong S, Rawls JF, Guillemin K et al. (2016). Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J 10: 655–664.
Daniels JA, Lederman HM, Maitra A, Montgomery EA . (2007). Gastrointestinal tract pathology in patients with common variable immunodeficiency (CVID). Am J Surg Pathol 31: 1800–1812.
Davenport ER, Cusanovich DA, Michelini K, Barreiro LB, Ober C, Gilad Y . (2015). Genome-wide association studies of the human gut microbiota. PLoS ONE 10: e0140301.
Dimitriu PA, Boyce G, Samarakoon A, Hartmann M, Johnson P, Mohn WW . (2013). Temporal stability of the mouse gut microbiota in relation to innate and adaptive immunity. Environ Microbiol Rep 5: 200–210.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Faith DP . (1992). Conservation evaluation and phylogenetic diversity. Biol Conserv 61: 1–10.
Flajnik MF, Kasahara M . (2010). Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat Rev Genet 11: 47–59.
Fox J, Weisberg S . (2011) An {R} Companion to Applied Regression. 2nd edition.Sage: Thousand Oaks, CA, USA.
Fransen F, Zagato E, Mazzini E, Fosso B, Manzari C, El Aidy S et al. (2015). BALB/c and C57BL/6 mice differ in polyreactive IgA abundance, which impacts the generation of antigen-specific IgA and microbiota diversity. Immunity 43: 527–540.
George LO, Bazzaz FA . (1999). The fern understory as an ecological filter: emergence and establishment of canopy-tree seedlings. Ecology 80: 833–845.
Goodrich JK, Davenport ER, Waters JL, Clark AG, Ley RE . (2016). Cross-species comparisons of host genetic associations with the microbiome. Science 352: 532–535.
Gori A, Tincati C, Rizzardini G, Torti C, Quirino T, Haarman M et al. (2008). Early impairment of gut function and gut flora supporting a role for alteration of gastrointestinal mucosa in human immunodeficiency virus pathogenesis. J Clin Microbiol 46: 757–758.
Hannon Lab. (2010).FASTX Toolkit. Available from http://hannonlab.cshl.edu/fastx_toolkit/index.html.
Hohn C, Petrie-Hanson L . (2012). Rag1−/− mutant zebrafish demonstrate specific protection following bacterial re-exposure. PLoS ONE 7: e44451.
Johansson MEV, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC . (2008). The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA 105: 15064–15069.
Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y et al. (2014). Foxp3+ T cells regulate immunoglobulin A selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 41: 152–165.
Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD et al. (2010). Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26: 1463–1464.
Kostic AD, Howitt MR, Garrett WS . (2013). Exploring host-microbiota interactions in animal models and humans. Genes Dev 27: 701–718.
Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, O’Connell RM et al. (2015a). MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe 17: 153–163.
Kubinak JL, Stephens WZ, Soto R, Petersen C, Chiaro T, Gogokhia L et al. (2015b). MHC variation sculpts individualized microbial communities that control susceptibility to enteric infection. Nat Commun 6: 8642.
Lam S, Chua HL, Gong Z, Lam TJ, Sin YM . (2004). Development and maturation of the immune system in zebrafish, Danio rerio: a gene expression profiling, in situ hybridization and immunological study. Dev Comp Immunol 28: 9–28.
Lau MK . (2013).DTK: Dunnett-Tukey-Kramer Pairwise Multiple Comparison Test Adjusted for Unequal Variances and Unequal Sample Sizes. Available from https://cran.r-project.org/package=DTK.
Love MI, Huber W, Anders S . (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550.
Lozupone C, Knight R . (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 8228–8235.
Magoc T, Salzberg SL . (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27: 2957–2963.
McMurdie PJ, Holmes S . (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8: e61217.
McMurdie PJ, Holmes S . (2014). Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol 10: e1003531.
Mouquet N, Loreau M . (2002). Coexistence in metacommunities: the regional similarity hypothesis. Am Nat 159: 420–426.
Oh J, Freeman AF, Park M, Sokolic R, Candotti F, Holland SM et al. (2013). The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res 23: 2103–2114.
Oksanen J, Blanchet FGG, Kindt R, Legendre P, Minchin PRR, O’Hara RBB et al. (2016).vegan: Community Ecology Package. Available from https://cran.r-project.org/package=vegan.
Org E, Parks BW, Joo JWJ, Emert B, Schwartzman W, Kang EY et al. (2015). Genetic and environmental control of host-gut microbiota interactions. Genome Res 25: 1558–1569.
Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L et al. (2014). Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158: 1000–1010.
R Core Team. (2015). R: A Language and Environment for Statistical Computing. Available from https://www.r-project.org/.
Rauta PR, Nayak B, Das S . (2012). Immune system and immune responses in fish and their role in comparative immunity study: a model for higher organisms. Immunol Lett 148: 23–33.
Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K et al. (2011). Evidence for a core gut microbiota in the zebrafish. ISME J 5: 1595–1608.
Shen W, Li W, Hixon JA, Bouladoux N, Belkaid Y, Dzutzev A et al. (2014). Adaptive immunity to murine skin commensals. Proc Natl Acad Sci USA 111: E2977–E2986.
Sloan WT, Lunn M, Woodcock S, Head IM, Nee S, Curtis TP . (2006). Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ Microbiol 8: 732–740.
Sørensen T . (1948). A method of establishing groups of equal amplitude in plant sociology based on similarity of species and its application to analysis of the vegetation on Danish commons. Biol Skr 5: 1–34.
Spor A, Koren O, Ley R . (2011). Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 9: 279–290.
Stephens WZ, Burns AR, Stagaman K, Wong S, Rawls JF, Guillemin K et al. (2016). The composition of the zebrafish intestinal microbial community varies across development. ISME J 10: 644–654.
Thoene-Reineke C, Fischer A, Friese C, Briesemeister D, Göbel UB, Kammertoens T et al. (2014). Composition of intestinal microbiota in immune-deficient mice kept in three different housing conditions. PLoS ONE 9: e113406.
Traver D, Herbomel P, Patton EE, Murphey RD, Yoder JA, Litman GW et al. (2003). The zebrafish as a model organism to study development of the immune system. Adv Immunol 81: 254–330.
Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI . (2008). Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3: 213–223.
Weinstein JA, Jiang N, White RA, Fisher DS, Quake SR . (2009). High-throughput sequencing of the zebrafish antibody repertoire. Science 324: 807–810.
Wickham H . (2009) ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag: New York, NY, USA.
Wienholds E . (2002). Target-selected inactivation of the zebrafish rag1 gene. Science 297: 99–102.
Wong S, Stephens WZ, Burns AR, Stagaman K, David LA, Bohannan BJM et al. (2015). Ontogenetic differences in dietary fat influence microbiota assembly in the zebrafish gut. MBio 6: e00687–15.
Zhang H, Sparks JB, Karyala SV, Settlage R, Luo XM . (2015). Host adaptive immunity alters gut microbiota. ISME J 9: 770–781.
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the NIH under award numbers R01GM095385 and P50GM098911. Grant P01HD22486 provided support for the Oregon Zebrafish Facility. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The ACISS computational resources were funded by a Major Research Instrumentation grant (Grant No. OCI-0960354) from the National Science Foundation, Office of Cyber Infrastructure. We also thank Rose Sockol for crossing and maintaining zebrafish in the Oregon Zebrafish Facility.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Stagaman, K., Burns, A., Guillemin, K. et al. The role of adaptive immunity as an ecological filter on the gut microbiota in zebrafish. ISME J 11, 1630–1639 (2017). https://doi.org/10.1038/ismej.2017.28
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.28
This article is cited by
-
Unravelling the temporal and spatial variation of fungal phylotypes from embryo to adult stages in Atlantic salmon
Scientific Reports (2024)
-
Collembolans maintain a core microbiome responding to diverse soil ecosystems
Soil Ecology Letters (2024)
-
The interactions between the host immunity and intestinal microorganisms in fish
Applied Microbiology and Biotechnology (2024)
-
Robust host source tracking building on the divergent and non-stochastic assembly of gut microbiomes in wild and farmed large yellow croaker
Microbiome (2022)
-
Host’s genetic background determines the outcome of reciprocal faecal transplantation on life-history traits and microbiome composition
Animal Microbiome (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}