
Abstract
Helicobacter suis is the second most prevalent Helicobacter species in the stomach of humans suffering from gastric disease. This bacterium mainly inhabits the stomach of domesticated pigs, in which it causes gastric disease, but it appears to be absent in wild boars. Interestingly, it also colonizes the stomach of asymptomatic rhesus and cynomolgus monkeys. The origin of modern human-, pig- or non-human primate-associated H. suis strains in these respective host populations was hitherto unknown. Here we show that H. suis in pigs possibly originates from non-human primates. Our data suggest that a host jump from macaques to pigs happened between 100 000 and 15 000 years ago and that pig domestication has had a significant impact on the spread of H. suis in the pig population, from where this pathogen occasionally infects humans. Thus, in contrast to our expectations, H. suis appears to have evolved in its main host in a completely different way than its close relative Helicobacter pylori in humans.
Similar content being viewed by others

Introduction
A number of Helicobacter species have been described to cause gastrointestinal disease in humans and animals. By far the best known and studied species is Helicobacter pylori, which can cause severe gastric disease in humans (Kusters et al., 2006). Humans are the major reservoir for H. pylori, although H. pylori infection in captive socially housed rhesus macaques also seems to be ubiquitous (Solnick et al., 2003). No evidence so far has been found on the presence of H. pylori in the stomach of chimpanzees, our closest living relatives (Moodley et al., 2012).
Besides H. pylori, other mainly animal-associated gastric Helicobacter species have been found colonizing the stomach of human patients, albeit at a much lower prevalence (Haesebrouck et al., 2009; Blaecher et al., 2013). These include dog- and cat-associated Helicobacter species, as well as H. suis. The latter commonly inhabits the stomach of domestic pigs and several studies in various countries on different continents have reported prevalences in pig herds exceeding 50% (Grasso et al., 1996; Park et al., 2004; Hellemans et al., 2007; Haesebrouck et al., 2009; Foss et al., 2013). Humans occasionally acquire the infection, most likely through direct contact with pigs or consumption of contaminated meat, and develop a gastritis which in some cases evolves to gastric MALT lymphoma (Stolte et al., 1997; Van den Bulck et al., 2005; Haesebrouck et al., 2009; De Cooman et al., 2013; Joosten et al., 2013). Direct human-to-human transmission of H. suis has not been reported so far. A strong correlation between H. suis infection and gastritis has also been observed in pigs (Grasso et al., 1996; De Bruyne et al., 2012) and conflicting evidence on the role of this bacterium in the development of ulceration of the keratinized pars oesophagea of the stomach has been reported (Grasso et al., 1996; Queiroz et al., 1996; Szeredi et al., 2005; De Bruyne et al., 2012). Furthermore, several studies attempting to detect this Helicobacter species in wild boars were unsuccessful (Fabisiak et al., 2010; Bassi, 2013), raising the question on the origin of H. suis infections in domesticated pigs.
H. suis has also been described to colonize rhesus monkeys (Macaca mulatta) and cynomolgus monkeys (crab-eating macaques; Macaca fascicularis) (O’Rourke et al., 2004; Martin et al., 2013; Bosschem et al., 2017). In contrast to what has been described in humans and pigs, H. suis infection in macaques in general seems asymptomatic (Drevon-Gaillot et al., 2006), although mild gastritis has been described occasionally (Dubois et al., 1991). All reports on H. suis infection in macaques relate to captive animals and, very often, a large number of animals from the studied groups are infected (Dubois et al., 1991; O’Rourke et al., 2004; Drevon-Gaillot et al., 2006; Nakamura et al., 2007; Martin et al., 2013). However, no data are available concerning the prevalence of H. suis in wild macaque populations, raising the question of a possible anthropogenic origin of H. suis infection in these animals.
The population structure and phylogeography of H. pylori have been shown to reflect human demographic events, such as ancient migrations of anatomically modern humans from Africa to the rest of the world (Falush et al., 2003; Moodley et al., 2009, 2012). These studies support the hypothesis that H. pylori co-evolved with its host for at least 100 000 years. For H. suis, however, no information is available on the worldwide phylogeny and at present, the origin of modern human-, pig- or non-human primate-associated Helicobacter suis strains in these respective host populations is unknown.
In the present study, we aimed at resolving the structure of the H. suis population colonizing pigs, humans and non-human primates, in order to investigate the true origin of the infection in these hosts. We show that H. suis in pigs possibly originates from non-human primates. Our data suggest that a host jump from macaques to pigs happened between 100 000 and 15 000 years ago and that pig domestication has had a significant impact on the spread of H. suis in the pig population, from where this pathogen occasionally infects humans.
Materials and methods
Samples
A list of the samples used for H. suis strain typing and their specifications is shown in Table 1 and Supplementary Table S1. Human samples were collected for diagnostic purposes during routine upper gastrointestinal tract endoscopy (O’Rourke et al., 2004; Joosten et al., 2013; Matsui et al., 2014). Stomachs from pigs (Sus scrofa) were collected in slaughterhouses or from wild boars that had been shot during authorized hunting. Gastric mucosal samples were collected as described previously (Liang et al., 2013). Tissues from captive cynomolgus monkeys, rhesus monkeys and mandrills from the USA, Japan and Australia had been previously collected and used for other purposes (Dubois et al., 1991; Lindén et al., 2004; O’Rourke et al., 2004; Nakamura et al., 2007; Martin et al., 2013).
In addition to tissues samples, seven and six H. suis strains were isolated using a previously described technique (Baele et al., 2008; Liang et al., 2015) from the stomach of socially housed rhesus monkeys (M. mulatta) and cynomolgus monkeys (M. fascicularis), respectively. These monkeys were bred and housed at the Biomedical Primate Research Centre (BPRC, Rijswijk, The Netherlands), accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). BPRC facilities comply with Dutch law on animal experiments and the EU Directive 63/2010, and have an Animal Welfare Assurance from NIH (A5539-01). Stomachs were collected at necropsy from healthy animals that were killed for reasons unrelated to this study.
All gastric tissue samples from pigs and humans, as well as in vitro isolated porcine strains described and used in the study by Liang et al. (2015) were included in the analyses as well.
Multilocus sequence typing
DNA from gastric biopsies as well as from pure H. suis strains was extracted using the Isolate Genomic DNA MiniKit (Bioline, London, UK) or DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer’s instructions. All DNA samples were screened for the presence of H. suis using a Taq polymerase-based species-specific PCR, as described by De Groote et al. (2000).
All in vitro isolated H. suis strains, as well as one tissue sample from every H. suis-positive animals and humans were subjected to multilocus sequence typing (MLST), as described previously (Liang et al., 2013). This technique allows culture-independent strain typing of fastidious microorganisms such as H. suis and is based on partial nucleotide sequencing of the following seven H. suis housekeeping genes: atpA, efp, ppa, mutY, trpC, ureAB and yphC.
Analysis of H. suis population structure, recombination analysis and estimation of time of divergence
The Maximum Composite Likelihood nucleotide distance was calculated based on the concatenated MLST sequences (4084 bp) using MEGA6 software (Tamura et al., 2004, 2013) and population structure and gene flow were inferred using Bayesian Analysis of the Population Structure (BAPS) 6 (Cheng et al., 2013).
To ensure that possible recombination events did not bias the phylogenetic analysis, two distinct approaches were used to analyze the sequences. First, ClonalFrame v1.2 was used to infer the ancestral relationship between H. suis strains analyzed in this study (Didelot and Falush, 2007). This software uses MLST data to estimate the clonal relationship between strains, while also taking into account the influence of horizontal gene transfer (Didelot and Falush, 2007; Moodley et al., 2012). The program was run two times with 500 000 Markov Chain Monte Carlo iterations with a thinning interval of 50 after an initial burn-in phase of 50 000 iterations and two times with 1 000 000 Markov Chain Monte Carlo iterations with a thinning interval of 100 after an initial burn-in phase of 100 000 iterations. A majority-rule consensus tree of the four independent ClonalFrame runs was computed and edited using the Interactive Tree of Life (iTOL) tool (Letunic and Bork, 2011). In a second approach, the method of Feil et al. (2003) was used to determine the pairwise nucleotide distances between strains as a function of MLST allele distances. Genes that have experienced recombination appeared as outliers in this analysis. The analysis was performed on the entire data set and for each BAPS cluster. After exclusion of genes affected by recombination, time of separation between the BAPS clusters HSU1 and HSU3 (see the Results section), showing a clear increasing linear tread in the analysis according to Feil and colleagues, was calculated using Bayesian Evolutionary Analysis by Sampling Trees (BEAST) (Drummond and Rambaut, 2007). A constant population size was assumed, and the HKY85 model of nucleotide substitution (Hasegawa et al., 1985) was used with strict clock and gamma rate heterogeneity with six categories (Drummond and Rambaut, 2007). A log-normal prior with a mean of 1.0 and s.d. of 1.25 on the logarithmic scale was assumed for the transition:tranversion ratio κ, and an exponential prior with a mean of 0.5 was utilized for the gamma shape parameter α. As prior, a normal distribution of the mutation rate ranging approximately between 1.8e−7 and 3.6e−7 (mean: 2.6e−7; s.d.: 8e−7; 95%, 1.28e−7–3.91e−7) was assumed, which was based on the long-term population-based mutation rate in the closely related human pathogen H. pylori (Moodley et al., 2009; Morelli et al., 2010). Linear regression, calculated using IBM SPSS statistics 23, was used to assess the strength of the correlation between the coalescent units estimated by Clonalframe and the time to the most recent common ancestor (TMRCA) estimated by BEAST, and to extrapolate the time of separation between the other BAPS clusters.
Determination of porcine mitochondrial DNA haplotype
Porcine gastric tissue was used to classify pigs as having a European or Asian haplotype, according to a previously described classification based on mitochondrial DNA (Larson et al., 2010). Briefly, PCR was performed in 20 μl reaction volumes containing 5U GoTaq Flexi DNA polymerase (Promega), 1 × GoTaq Flexi PCR buffer, 2.5 mm MgCl2 (Promega), 200 μm dNTPs (Bioline), 0.25 μm of both primers (DloopL: 5′-ACTAACTCCGCCATCAGCAC-3′; DloopR: 5′-GTTTGGCAAGGCGTTATAG-3′) and 1 μl DNA. PCR reactions were run in a MasterCycler thermal cycler (Eppendorf) using the following cycling conditions: initial denaturation for 3 min at 94 °C, followed by 35 cycles of 94 °C for 1 min, 58 °C for 1 min, 72 °C for 1 min and a final extension of 10 min at 72 °C. Alternatively, a newly designed primer pair was used to amplify and sequence part of the mitochondrial control region (D-loop; F: 5′-CTCGCTCCGGGCCCATAA-3′; R: 5′-TTTTTGGGGTTTGGCAA-3′). Based on the obtained sequences (Supplementary Figure S1), animals were classified as having a European or Asian haplotype, according to the classification described by Larson et al. (2005) (Supplementary Table S1).
The circos software package (Krzywinski et al., 2009) was used to visualize the distribution of the H. suis BAPS clusters among the different host species and genotypes (i.e. Homo sapiens, M. fascicularis, M. mulatta, Mandrillus sphinx, Asian S. scrofa, European S. scrofa).
Results
For 56 porcine samples out of a total of 80 for which a piece of gastric tissue was available, we were able to determine the mitochondrial DNA haplotype as being European or Asian (Supplementary Table S1 and Supplementary Figure S1). All Chinese and Vietnamese pigs were shown to have an Asian haplotype. Most pigs from Europe and the Americas revealed a European haplotype, although some of these animals revealed an Asian haplotype.
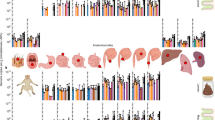
A total of 149 H. suis-positive samples from domestic pigs, wild boars, cynomolgus monkeys, rhesus monkeys and humans were collected and subjected to MLST. All obtained sequences are available at http://pubmlst.org/hsuis/. ClonalFrame and BAPS analysis revealed that H. suis strains clearly belong to five distinct populations (Figures 1a and 2 and Supplementary Table S1). Population HSU3 includes all H. suis strains isolated from or detected in cynomolgus monkeys. Populations HSU2 and HSU4 contain all strains colonizing the stomach of rhesus monkeys, as well as the two strains from the mandrills. The fact that no distinction could be made between H. suis strains from rhesus monkeys and mandrills, while both primate species originate from a different continent, suggests that the two mandrills harboring H. suis may have contracted the infection through direct or indirect contact with or the proximity of rhesus monkeys in the same Australian zoo.

Helicobacter suis population structure. ClonalFrame and BAPS analysis showed that H. suis strains clearly belong to five distinct populations (HSU1–HSU5) (a). Population HSU5 was further divided into four subpopulations (HSU5_1–HSU5_4) (b). Gene flow between different H. suis BAPS populations (c) and HSU5 subpopulations (d) are indicated by arrows, accompanied by a number between 0 and 1, representing the proportion of ancestral DNA flow in this direction. Colors in Figures 1a and b correspond to the colors attributed to different BAPS clusters in Figures 1c and d, respectively. An * designates H. suis strains showing a mixed ancestry, as shown by mixed bar colors.

Association between the population structure of the host and that of H. suis. This figure shows the distribution of the H. suis strains from different BAPS clusters (HSU1–HSU5) among the different host species and genotypes (i.e. Homo sapiens, Macaca fascicularis, Macaca mulatta, Mandrillus sphinx, Asian Sus scrofa, European Sus scrofa).
Populations HSU1 and HSU5 comprise all the strains detected in or isolated from pigs and humans. More specifically, HSU1 (referred to as the ‘Asian’ pig-associated population) contains most, but not all, of the strains colonizing pigs with Asian mitochondrial DNA (mtDNA) haplotypes originating from Vietnam and China, while population HSU5 (referred to as the ‘Euroasian’ pig- and human-associated H. suis population) comprises all strains colonizing pigs with European haplotypes (originating from a European, American or African host), some strains colonizing pigs with Asian haplotypes (both originating from Europe and Asia) as well as all H. suis strains colonizing humans (Supplementary Table S1). This, along with the relatively low prevalence of H. suis infection in humans, suggests that H. suis bacteria occasionally jump from pigs to humans. Finally, population HSU5 was further divided (Figure 1b) into HSU5_1 and HSU5_2, harboring H. suis strains colonizing pigs with European or Asian haplotypes, and HSU5_3 and HSU5_4, comprising strains circulating only in pigs showing Asian haplotypes. Overall, a clear association was observed between the population structure of the host and that of H. suis (Figure 2).
Admixture analysis revealed gene flow between the different BAPS clusters, particularly from H. suis populations circulating in both macaque species (HSU2 and HSU3) to the pig- (and human)-associated Euroasian population (HSU5) and between the two H. suis populations circulating in pigs (HSU_1 and HSU_5) (Figure 1c). In addition, gene flow between Euroasian and Asian strains was also inferred within the HSU5 population (Figure 1d).
The common ancestor of the pig-associated H. suis population was shown to lie within the diversity of all macaque-associated H. suis, with the cynomolgus monkey-associated helicobacters being the sister clade of pig-associated H. suis strains (Figure 1a). Despite the higher number of pig- and human-associated H. suis strains analyzed in this study, the diversity of H. suis strains in non-human primates appeared to be much greater compared with the diversity of H. suis strains circulating in humans and pigs, which showed a star-like phylogeny characterized by long internal and short external tree branches. This strongly indicates a more ancient association between H. suis and non-human primates compared with that between H. suis and pigs, as well as a possible recent clonal expansion of the H. suis population colonizing domesticated pigs worldwide.
Nucleotide distance analysis revealed an identity of approximately 95% (94.4–95.15%) between the different H. suis populations from monkeys and pigs, whereas the highest distance (93% identity) was observed between the two groups harboring macaque-associated H. suis strains.
Using BEAST, we estimated the age of the association between H. suis and its hosts, based on the described population-based mutation rate for the closely related species H. pylori (Table 2). To minimize the effect of recombination in the estimation, the yphC gene was excluded from the analysis, since this was shown to be affected by pervasive recombination. In addition, we limited the BEAST analysis to populations HSU1 (H. suis strains associated with Asian pigs) and HSU3 (H. suis strains found in M. fascicularis). These two populations were selected since they showed a clear increasing linear tread in the analysis according to Feil et al. (2003), indicating little effect of recombination on the evolution of these lineages. Medians and the 95% highest posterior density interval of the estimation of the time to the most recent common ancestor (TMRCA) calculated by BEAST are summarized in Table 2. Furthermore, we attempted to infer the ages of the other nodes in the ClonalFrame tree by applying linear regression between the coalescent time calculated by Clonalframe and the log10 of the TMRCA inferred by BEAST. A strong correlation was observed (R2=0.942) and the predicted ages of the other nodes are listed in Table 2. We estimated that a common ancestor for the H. suis population in pigs and monkeys existed until approximately 200 000 (193 118–197 704) years ago. In addition, we dated the split between monkey-associated and pig-associated H. suis populations approximately 100 000 (146 650–71 649) years ago. The H. suis population in pigs underwent a subsequent clonal expansion starting approximately 15 000 years ago. This, together with the limited diversity in the pig-associated H. suis population and the mixed ancestry (Figure 1a) observed for several pig-associated H. suis strains, strongly suggests that H. suis in pigs originates from non-human primates, a jump which seems to have taken place somewhere between 100 000 and 15 000 years ago.
Discussion
Numerous studies performed in Europe, the Americas and Asia have shown that H. suis infection in pigs is very common (Grasso et al., 1996; Park et al., 2004; Hellemans et al., 2007; Foss et al., 2013) with prevalences exceeding 50% and often being as high as 80–90%. These numbers resemble the high prevalence of H. pylori infection in humans worldwide, leading to the hypothesis that H. suis has evolved in pigs in a similar way to what has been described for H. pylori in humans. Several studies have suggested a long-standing coevolution of H. pylori and humans for at least 100 000 years, resulting in a large genetic diversity and relatively strong phylogeographic signals (Achtman et al., 1999; Falush et al., 2003; Moodley et al., 2009). In contrast, the results of the present study revealed that the H. suis population in pigs shows a fairly limited genetic diversity. In addition, the main pig-associated H. suis population (HSU5) showed a star-like phylogeny with relatively few structured geographic signals. The clearest distinction was found between approximately half of the H. suis strains colonizing Asian pigs and all other H. suis strains, associated with pigs in Europe, the Americas and Africa as well as infected humans from different continents. In contrast and somewhat surprisingly, the H. suis populations colonizing rhesus and cynomolgus monkeys could clearly be subdivided into two clades with a much greater genetic diversity. This very strongly suggests a more ancient association between H. suis and non-human primates than the association between S. scrofa and H. suis, and it appears to disprove the original idea of the swine origin of H. suis. In addition, for some pig-associated H. suis strains, a mixed ancestry was observed with the population colonizing cynomolgus monkeys indicating possible gene flow between these populations. This further indicates that H. suis infection in the pig population worldwide may very well originate from non-human primates. This could, for instance, have taken place through the consumption of monkey stomachs or their contents by pigs, which are indeed considered to be omnivorous. However, we cannot exclude other possible scenarios including the involvement of a so far unknown intermediate host species of H. suis or the (near-)extinction of the original pig-associated H. suis population due to a severe bottleneck as observed in other pathogens (Kapusinszky et al., 2015). In our case, the domestication of S. scrofa and subsequent selection for specific traits may have caused a significant bottleneck effect on the diversity of pig-associated H. suis originally circulating in the ancestral wild boar population. This hypothesis implies the possibility that specific H. suis strains still colonize wild boars around the world. However, this theory is contradicted by several studies which unsuccessfully attempted to detect H. suis in these animals (Fabisiak et al., 2010; Bassi, 2013).
In the present study, samples from Belgian and Chinese wild boars were also tested for the presence of H. suis DNA. The two Chinese animals were negative for H. suis, as well as 7/9 Belgian wild boars (data not shown). The two positive animals only revealed the presence of very low numbers of H. suis in the cardiac gland zone or the pars oesophagea, which have been described not to be the main colonization sites for this bacterium in pigs (Hellemans et al., 2007; De Bruyne et al., 2012). Both strains belong to the ‘Euroasian’ pig- and human-associated H. suis population, so detection of H. suis DNA in atypical stomach regions in these animals might be a consequence of recent contamination, for instance through contact with domesticated animals or their excretes. In general, our results are consistent with previous studies revealing that H. suis seems to be absent in the majority of the wild boar populations (Fabisiak et al., 2010; Bassi, 2013). This further supports our theory of a fairly recent host jump of H. suis to pigs from macaques or other unknown hosts, although further research should be performed in additional wild boar populations around the globe to provide more insights. Recently, a novel Helicobacter species, H. apri, has been described in wild boars, but this is an enterohepatic Helicobacter species that does not produce urease (Zanoni et al., 2016). It was, therefore, not included in our study.
An estimation of the divergence time between the different clades suggests that the host jump from macaques to pigs may have taken place somewhere between 100 000 years ago (ya), the inferred date of the split between monkey- and pig-associated populations, and 15 000 ya, which is the time to the most recent common ancestor of pig-associated H. suis populations. This would imply that pigs got infected before the domestication of this animal species. Both zooarcheological and molecular studies have revealed at least two centers of pig domestication, which clearly happened independently in the Near East (Anatolia) and East Asia (Mekong valley) around 9000 ya (Larson et al., 2005; Cucchi et al., 2011; Frantz et al., 2015). The first domestic pigs in Europe were most likely dispersed from Anatolia into Europe around 7500 ya, forming the basis for western Eurasian domestic pigs (Larson et al., 2007). Nevertheless, recent data have revealed a more complex domestication process involving continuous post-domestication gene flow between domestic pig and wild boar populations, both in Asia and Europe, leading to the loss of Near Eastern mtDNA signatures in European domesticated pigs (Larson et al., 2007; Frantz et al., 2015). Our data showed that the host jump from H. suis was followed by a clonal expansion starting ~15 000 ya, suggesting that domestication of S. scrofa 7000–9000 ya and selection post-domestication for behavioral and morphological traits (Frantz et al., 2015) may have had an significant impact on the spread of this bacterium in the modern pig population.
The results of the present study do not allow an exact calculation of the age of the association between H. suis and macaques. Possibly, the ancestor of M. fascicularis and M. mulatta, which have separated ~2.5 Mya, was infected with an ancestor of modern H. suis, or alternatively, one macaque species served as a source of infection for the other species, for instance during periods of interbreeding that have been described to have taken place after approximately 170 000 before the present in Indochina (Tosi et al., 2002; Kanthaswamy et al., 2008). As mentioned above, for some pig-associated H. suis strains a mixed ancestry was observed with the clade harboring all cynomolgus monkey-associated H. suis strains. The latter was also shown to be the sister clade of pig-associated H. suis strains, showing that cynomolgus monkeys are the most likely ancestral source of H. suis infection in pigs, rather than rhesus macaques, unless an unknown intermediate host species is involved as well. Bearing in mind the natural habitat of the animals (Kanthaswamy et al., 2008), a potential host jump of H. suis from either macaque species to pigs most likely would have happened in East Asia. In turn, East Asian pigs may have been the source of H. suis infection in western domesticated pigs. Indeed, Asian pigs, possibly harboring H. suis, were introduced into Europe during the eighteenth and nineteenth centuries, as shown by historical records and recent investigations. These have revealed an Asian introgression of about 20% into the genome of European commercial pigs (Bosse et al., 2014), as well as the presence of a minority of Asian mtDNA haplotypes in several European and American pig breeds (Giuffra et al., 2000; Larson et al., 2010), which was also demonstrated in the present study.
In our study, we clearly demonstrated differences between H. suis strains isolated from pigs and both macaque species. Similar reports have been made for other pathogens that have jumped between different hosts, such as Staphylococcus aureus (Smith et al., 2014). It is of course important to realize that not only the pathogen but also the interaction between the host and pathogen determines the outcome of an infection. This has been nicely demonstrated for the closely related human pathogen H. pylori. African H. pylori ancestry, for instance, has been shown to be relatively benign in humans from African origin, whereas it is deleterious in individuals with substantial Amerindian ancestry, showing that the disruption of coevolution, as is the case for Amerindian humans infected with an African H. pylori strain, may result in more severe pathology (Kodaman et al., 2014). Another example of disease severity associated with disrupted coevolution is that of bovine papillomavirus-1 (BPV-1), which causes relatively innocent warts in cows, its natural host. Recent research has revealed evidence of multiple, relatively recent host jumps of BPV-1 into horses, in which this virus can cause the development of invasive and aggressive sarcoids (Trewby et al., 2014). In the present study, we revealed a more ancient association between H. suis and macaques, compared with that between H. suis and pigs. The seemingly more innocent character of H. suis infection in macaques fits with the examples described above for H. pylori and the bovine papillomavirus-1, showing that coevolution between the microbe and the host generally results in decreased pathogenicity. In contrast, the fairly recent host jump and expansion of a clonal population, especially predicted for the European domestic pig population, may explain the pathogenicity of H. suis strains infecting pigs.
This study provides the first insights into the phylogeny and population structure of H. suis. In contrast to our expectations, we showed that H. suis in pigs probably originates from non-human primates. After a possible host jump from macaques to pigs, pig domestication may have had a significant impact on the spread of H. suis in the pig population, from where this pathogen occasionally infects humans. This is a rare example showing that an important microbial pathogen in pigs and humans in fact seems to originate from a completely different host species, in which infection causes little or no harm. Future research should aim to answer the question whether other, hitherto unknown host species may have contributed to the evolution and worldwide spread of H. suis. In addition, it should be investigated which adaptations may have facilitated the remarkable jump of H. suis from non-human primates to pigs and the subsequent spread of the microorganism in its new host.
References
Achtman M, Azuma T, Berg DE, Ito Y, Morelli G, Pan ZJ et al. (1999). Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol Microbiol 32: 459–470.
Baele M, Decostere A, Vandamme P, Ceelen L, Hellemans A, Mast J et al. (2008). Isolation and characterization of Helicobacter suis sp. nov. from pig stomachs. Int J Syst Evol Microbiol 58: 1350–1358.
Bassi P . (2013). Rilievi anatomo-patologici e batteriologici condotti sull’apparato gastroenterico di una metapopolazione di cinghiali (Sus scrofa, Alma Mater Studiorum Università di Bologna. Dottorato di ricerca in Morfofisiologia e patologia veterinaria con applicazioni biotecnologiche, 25 Ciclo, Dissertation thesis. doi: 10.6092/unibo/amsdottorato/5518. http://amsdottorato.unibo.it/5518/.
Blaecher C, Smet A, Flahou B, Pasmans F, Ducatelle R, Taylor D et al. (2013). Significantly higher frequency of Helicobacter suis in patients with idiopathic parkinsonism than in control patients. Aliment Pharmacol Ther 38: 1347–1353.
Bosschem I, Flahou B, Bakker J, Heuvelman E, Langermans JAM, De Bruyne E et al. (2017). Comparative virulence of in vitro-cultured primate- and pig-associated Helicobacter suis strains in a BALB/c mouse and Mongolian gerbil model. Helicobacter 22: e12349.
Bosse M, Madsen O, Megens HJ, Frantz LA, Paudel Y, Crooijmans RP et al. (2014). Hybrid origin of European commercial pigs examined by an in-depth haplotype analysis on chromosome 1. Front Genet 5: 442.
Cheng L, Connor TR, Siren J, Aanensen DM, Corander J . (2013). Hierarchical and spatially explicit clustering of DNA sequences with BAPS software. Mol Biol Evol 30: 1224–1228.
Cucchi T, Hulme-Beaman A, Yuan J, Dobney K . (2011). Early Neolithic pig domestication at Jiahu, Henan Province, China: clues from molar shape analyses using geometric morphometric approaches. J Archaeol Sci 38: 11–22.
De Bruyne E, Flahou B, Chiers K, Meyns T, Kumar S, Vermoote M et al. (2012). An experimental Helicobacter suis infection causes gastritis and reduced daily weight gain in pigs. Vet Microbiol 160: 449–454.
De Cooman L, Flahou B, Houf K, Smet A, Ducatelle R, Pasmans F et al. (2013). Survival of Helicobacter suis bacteria in retail pig meat. Int J Food Microbiol 166: 164–167.
De Groote D, Ducatelle R, van Doorn L, Tilmant K, Verschuuren A, Haesebrouck F . (2000). Detection of "Candidatus Helicobacter suis" in gastric samples of pigs by PCR: comparison with other invasive diagnostic techniques. J Clin Microbiol 38: 1131–1135.
Didelot X, Falush D . (2007). Inference of bacterial microevolution using multilocus sequence data. Genetics 175: 1251–1266.
Drevon-Gaillot E, Perron-Lepage M-F, Clement C, Burnett R . (2006). A review of background findings in cynomolgus monkeys (Macaca fascicularis from three different geographical origins. Exp Toxicol Pathol 58: 77–88.
Drummond AJ, Rambaut A . (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7: 214.
Dubois A, Tarnawski A, Newell DG, Fiala N, Dabros W, Stachura J et al. (1991). Gastric injury and invasion of parietal cells by spiral bacteria in rhesus monkeys. Are gastritis and hyperchlorhydria infectious diseases? Gastroenterology 100: 884–891.
Fabisiak M, Sapierzynski R, Salamaszynska-Guz A, Kizerwetter-Swida M . (2010). The first description of gastric Helicobacter in free-ranging wild boar (Sus scrofa from Poland. Pol J Vet Sci 13: 171–174.
Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M et al. (2003). Traces of human migrations in Helicobacter pylori populations. Science 299: 1582–1585.
Feil EJ, Cooper JE, Grundmann H, Robinson DA, Enright MC, Berendt T et al. (2003). How clonal is Staphylococcus aureus? J Bacteriol 185: 3307–3316.
Foss D, Kopta L, Paquette J, Bowersock T, Choromanski L, Galvin J et al. (2013). Identification of Helicobacter suis in pig-producing regions of the United States. J Swine Health Prod 21: 242–247.
Frantz LA, Schraiber JG, Madsen O, Megens HJ, Cagan A, Bosse M et al. (2015). Evidence of long-term gene flow and selection during domestication from analyses of Eurasian wild and domestic pig genomes. Nat Genet 47: 1141–1148.
Giuffra E, Kijas JMH, Amarger V, Carlborg O, Jeon JT, Andersson L et al. (2000). The origin of the domestic pig: independent domestication and subsequent introgression. Genetics 154: 1785–1791.
Grasso G, Ripabelli G, Sammarco M, Ruberto A, Iannitto G . (1996). Prevalence of Helicobacter-like organisms in porcine gastric mucosa: a study of swine slaughtered in Italy. Comp Immunol Microbiol Infect Dis 19: 213–217.
Haesebrouck F, Pasmans F, Flahou B, Chiers K, Baele M, Meyns T et al. (2009). Gastric helicobacters in domestic animals and nonhuman primates and their significance for human health. Clin Microbiol Rev 22: 202–223.
Hasegawa M, Kishino H, Yano TA . (1985). Dating of the human ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22: 160–174.
Hellemans A, Chiers K, Maes D, De Bock M, Decostere A, Haesebrouck F et al. (2007). Prevalence of 'Candidatus Helicobacter suis' in pigs of different ages. Vet Rec 161: 189–192.
Joosten M, Flahou B, Meyns T, Smet A, Arts J, De Cooman L et al. (2013). Case report: Helicobacter suis infection in a pig veterinarian. Helicobacter 18: 392–396.
Kanthaswamy S, Satkoski J, George D, Kou A, Erickson BJ-A, Smith DG et al. (2008). Hybridization and stratification of nuclear genetic variation in Macaca mulatta and M. fascicularis. Int J Primatol 29: 1295–1311.
Kapusinszky B, Mulvaney U, Jasinska AJ, Deng X, Freimer N, Delwart E et al. (2015). Local virus extinctions following a host population bottleneck. J Virol 89: 8152–8161.
Kodaman N, Pazos A, Schneider BG, Piazuelo MB, Mera R, Sobota RS et al. (2014). Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci USA 111: 1455–1460.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645.
Kusters JG, van Vliet AHM, Kuipers EJ . (2006). Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev 19: 449–490.
Larson G, Dobney K, Albarella U, Fang MY, Matisoo-Smith E, Robins J et al. (2005). Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Science 307: 1618–1621.
Larson G, Albarella U, Dobney K, Rowley-Conwy P, Schibler J, Tresset A et al. (2007). Ancient DNA, pig domestication, and the spread of the Neolithic into Europe. Proc Natl Acad Sci USA 104: 15276–15281.
Larson G, Liu R, Zhao X, Yuan J, Fuller D, Barton L et al. (2010). Patterns of East Asian pig domestication, migration, and turnover revealed by modern and ancient DNA. Proc Natl Acad Sci USA 107: 7686–7691.
Letunic I, Bork P . (2011). Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39: W475–W478.
Liang J, Ducatelle R, Pasmans F, Smet A, Haesebrouck F, Flahou B . (2013). Multilocus sequence typing of the porcine and human gastric pathogen Helicobacter suis. J Clin Microbiol 51: 920–926.
Liang J, De Bruyne E, Ducatelle R, Smet A, Haesebrouck F, Flahou B . (2015). Purification of Helicobacter suis strains from biphasic cultures by single colony isolation: influence on strain characteristics. Helicobacter 20: 206–216.
Lindén S, Boren T, Dubois A, Carlstedt I . (2004). Rhesus monkey gastric mucins: oligomeric structure, glycoforms and Helicobacter pylori binding. Biochem J 379: 765–775.
Martin M, Bhatnagar S, George M, Paster B, Canfield D, Eisen J et al. (2013). The impact of Helicobacter pylori infection on the gastric microbiota of the rhesus macaque. PLoS ONE 8: e76375.
Matsui H, Takahashi T, Murayama SY, Uchiyama I, Yamaguchi K, Shigenobu S et al. (2014). Development of new PCR primers by comparative genomics for the detection of Helicobacter suis in gastric biopsy specimens. Helicobacter 19: 260–271.
Moodley Y, Linz B, Yamaoka Y, Windsor HM, Breurec S, Wu JY et al. (2009). The peopling of the Pacific from a bacterial perspective. Science 323: 527–530.
Moodley Y, Linz B, Bond RP, Nieuwoudt M, Soodyall H, Schlebusch CM et al. (2012). Age of the association between Helicobacter pylori and Man. PLoS Pathog 8: e1002693.
Morelli G, Didelot X, Kusecek B, Schwarz S, Bahlawane C, Falush D et al. (2010). Microevolution of Helicobacter pylori during prolonged infection of single hosts and within families. PLoS Genet 6: e1001036.
Nakamura M, Murayama SY, Serizawa H, Sekiya Y, Eguchi M, Takahashi S et al. (2007). ‘Candidatus Helicobacter heilmannii’ from a cynomolgus monkey induces gastric mucosa-associated lymphoid tissue lymphomas in C57BL/6 mice. Infect Immun 75: 1214–1222.
O'Rourke J, Solnick J, Neilan B, Seidel K, Hayter R, Hansen L et al. (2004). Description of 'Candidatus Helicobacter heilmannii' based on DNA sequence analysis of 16 S rRNA and urease genes. Int J Syst Evol Microbiol 54: 2203–2211.
Park J, Seok S, Cho S, Baek M, Lee H, Kim D . (2004). The high prevalence of Helicobacter sp. in porcine pyloric mucosa and its histopathological and molecular characteristics. Vet Microbiol 104: 219–225.
Queiroz DMD, Rocha GA, Mendes EN, deMoura SB, deOliveira AMR, Miranda D . (1996). Association between Helicobacter and gastric ulcer disease of the pars esophagea in swine. Gastroenterology 111: 19–27.
Smith EM, Needs PF, Manley G, Green LE . (2014). Global distribution and diversity of ovine-associated Staphylococcus aureus. Infect Genet Evol 22: 208–215.
Solnick JV, Chang K, Canfield DR, Parsonnet J . (2003). Natural acquisition of Helicobacter pylori infection in newborn rhesus Macaques. J Clin Microbiol 41: 5511–5516.
Stolte M, Kroher G, Meining A, Morgner A, Bayerdörffer E, Bethke B . (1997). Comparison of Helicobacter pylori and H. heilmannii gastritis. A matched control study involving 404 patients. Scand J Gastroenterol 32: 28–33.
Szeredi L, Palkovics G, Solymosi N, Tekes L, Mehesfalvi J . (2005). Study on the role of gastric Helicobacter infection in gross pathological and histological lesions of the stomach in finishing pigs. Acta Vet Hung 53: 371–383.
Tamura K, Nei M, Kumar S . (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA 101: 11030–11035.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S . (2013). MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol 30: 2725–2729.
Tosi AJ, Morales JC, Melnick DJ . (2002). Y-chromosome and mitochondrial markers in Macaca fascicularis indicate introgression with Indochinese M. mulatta and a biogeographic barrier in the Isthmus of Kra. Int J Primatol 23: 161–178.
Trewby H, Ayele G, Borzacchiello G, Brandt S, Campo MS, Del Fava C et al. (2014). Analysis of the long control region of bovine papillomavirus type 1 associated with sarcoids in equine hosts indicates multiple cross-species transmission events and phylogeographical structure. J Gen Virol 95: 2748–2756.
Van den Bulck K, Decostere A, Baele M, Driessen A, Debongnie JC, Burette A et al. (2005). Identification of non-Helicobacter pylori spiral organisms in gastric samples from humans, dogs, and cats. J Clin Microbiol 43: 2256–2260.
Zanoni RG, Piva S, Florio D, Bassi P, Mion D, Cnockaert M et al. (2016). Helicobacter apri sp. nov., isolated from wild boars. Int J Syst Evol Microbiol 66: 2876–2882.
Acknowledgements
This study was funded by a grant from the Research Fund of Ghent University, Ghent, Belgium (grant BOF14/GOA/010) and the Research Foundation—Flanders (FWO; grant FWO14/PDO/067). JC was funded by the COIN centre of excellence, Academy of Finland grant 2551170. SKL was funded by grants from the Swedish Research Council (grant 521-2011-2370) and Formas (grant 221-2013-590).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Flahou, B., Rossi, M., Bakker, J. et al. Evidence for a primate origin of zoonotic Helicobacter suis colonizing domesticated pigs. ISME J 12, 77–86 (2018). https://doi.org/10.1038/ismej.2017.145
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.145
This article is cited by
-
Cross-species single-cell transcriptomic analysis of animal gastric antrum reveals intense porcine mucosal immunity
Cell Regeneration (2023)
-
Molecular detection of Helicobacter spp. and Fusobacterium gastrosuis in pigs and wild boars and its association with gastric histopathological alterations
Veterinary Research (2022)
-
Gastric Helicobacter species associated with dogs, cats and pigs: significance for public and animal health
Veterinary Research (2022)
-
Antibiotic resistance pattern and frequency of cagA and vacA genes in Helicobacter pylori strains isolated from patients in Tabriz city, Iran
BMC Research Notes (2021)
-
Distinct transcriptome signatures of Helicobacter suis and Helicobacter heilmannii strains upon adherence to human gastric epithelial cells
Veterinary Research (2020)
