
Abstract
The recently isolated strain L21-Fru-ABT represents moderately halophilic, obligately anaerobic and saccharolytic bacteria that thrive in the suboxic transition zones of hypersaline microbial mats. Phylogenetic analyses based on 16S rRNA genes, RpoB proteins and gene content indicated that strain L21-Fru-ABT represents a novel species and genus affiliated with a distinct phylum-level lineage originally designated Verrucomicrobia subdivision 5. A survey of environmental 16S rRNA gene sequences revealed that members of this newly recognized phylum are wide-spread and ecologically important in various anoxic environments ranging from hypersaline sediments to wastewater and the intestine of animals. Characteristic phenotypic traits of the novel strain included the formation of extracellular polymeric substances, a Gram-negative cell wall containing peptidoglycan and the absence of odd-numbered cellular fatty acids. Unusual metabolic features deduced from analysis of the genome sequence were the production of sucrose as osmoprotectant, an atypical glycolytic pathway lacking pyruvate kinase and the synthesis of isoprenoids via mevalonate. On the basis of the analyses of phenotypic, genomic and environmental data, it is proposed that strain L21-Fru-ABT and related bacteria are specifically adapted to the utilization of sulfated glycopolymers produced in microbial mats or biofilms.
Similar content being viewed by others


Introduction
The bacterial phyla Planctomycetes, Verrucomicrobia, Chlamydiae, Lentisphaerae and Omnitrophica form a monophyletic group in most phylogenetic trees and thus probably share a common ancestor. This assemblage is commonly referred to as the PVC superphylum after the initial letters of the three most well-known phyla. Originally, members of the proposed phylum Poribacteria so far mainly found in sponges (Fieseler et al., 2004) were also included in this superphylum (Wagner and Horn, 2006). However, recent phylogenetic analyses indicate that Poribacteria form a separate lineage that does not associate with representatives of the PVC superphylum (Gupta et al., 2012; Kamke et al., 2014). The five currently recognized phyla of the PVC superphylum comprise several groups of bacteria with a high relevance in basic research (for example, Santarella-Mellwig et al., 2013; Jeske et al., 2015), biotechnology (for example, Jetten et al., 2001) and medicine (for example, Horn et al., 2004). In addition, some environmental clades of the Verrucomicrobia phylum can be of high ecological significance, especially in soils where their abundance can exceed 20% of the total bacterial community (Bergmann et al., 2011).
Despite their importance in ecology, the portion of culturable bacteria within the Verrucomicrobia is quite low compared with the observed phylogenetic breadth (Hugenholtz et al., 1998). In particular, a group of 16S rRNA sequences tentatively defined as subdivision 5 of Verrucomicrobia by Hugenholtz et al. (1998) is still lacking a described species, although cultivation-independent studies reported a wide-spread and abundant occurrence in marine environments (Cardman et al., 2014) and the intestinal tract of several vertebrates (Frey et al., 2006; Steelman et al., 2012). The first pure culture representing this phylogenetic group was recently isolated from an anoxic cyanobacterial mat sample retrieved from a hypersaline lake on the Kiritimati Atoll (Spring et al., 2015). In this study, we present a detailed description of the phenotypic traits and complete genome sequence of the novel strain L21-Fru-ABT that allows for the first time the reconstruction of the metabolic potential and lifestyle of a member of the tentatively defined subdivision 5. On the basis of the description of strain L21-Fru-ABT, the novel species and genus Kiritimatiella glycovorans is proposed as well as several novel taxa of higher ranks including the novel phylum Kiritimatiellaeota comprising environmental sequences allocated previously to the Verrucomicrobia subdivision 5.
Materials and methods
Strains and cultivation conditions
Strain L21-Fru-ABT was enriched from an anoxic sample of a cyanobacterial mat obtained from the hypersaline Lake 21 located on the atoll of Kiritimati (Republic of Kiribati, Central Pacific) and deposited in the Leibniz Institute German Collection of Microorganisms and Cell Cultures (DSMZ; Braunschweig, Germany) and Japan Collection of Microorganisms (JCM; Tsukuba, Japan) under the accession numbers DSM 26986T and JCM 19195T, respectively. A detailed description of the sample location was provided previously (Schneider et al., 2013; Spring et al., 2015) and further details on the methods for the enrichment and isolation of this strain are given by Spring et al. (2015). For the preparation of media and incubation under anoxic conditions, the anaerobe cultivation technique of Hungate (1950) with the modifications introduced by Bryant (1972) was used. The basal medium for the characterization of strain L21-Fru-ABT contained per litre: 60.0 g NaCl, 6.0 g MgCl2 × 6 H2O, 1.5 g KCl, 1.0 g Na2SO4, 1.0 g NH4Cl, 0.4 g CaCl2 × 2 H2O, 0.4 g K2HPO4, 10.0 ml trace elements solution of DSMZ medium 141 (http://www.dsmz.de/microorganisms/medium/pdf/DSMZ_Medium141.pdf), 0.5 mg resazurin, 10.0 ml vitamins solution of DSMZ medium 141, 2.5 g Na2CO3, 0.3 g Na2S × 9 H2O and 0.3 g l-cysteine-HCl × H2O. The medium was prepared under 80% N2 and 20% CO2 gas mixture without the vitamins, carbonate, sulfide and cysteine, which were added to the medium after autoclaving from sterile anoxic stock solutions. For routine cultivation, 1.0 g l−1 d-glucose was used as substrate. The pH of the completed medium was adjusted to 7.3–7.5 and the standard incubation temperature was 28 °C.
For comparative analyses of the cellular fatty acid patterns and diagnostic diamino acids of peptidoglycan, the following strains were obtained from the Leibniz Institute DSMZ: Escherichia coli DSM 498, Opitutus terrae DSM 11246T, Victivallis vadensis DSM 14823T, Akkermansia muciniphila DSM 22959T, Spirochaeta dissipatitropha DSM 23605T and Oligosphaera ethanolica DSM 24202T. For cultivation of these strains, the medium recipes and incubation conditions indicated in the catalog of the Leibniz Institute DSMZ (http://www.dsmz.de/catalogues/catalogue-microorganisms.html) were used. Single compounds were obtained from Sigma-Aldrich (Taufkirchen, Germany) and complex nutrients from BD Biosciences (Heidelberg, Germany).
Genome sequencing and analyses
Genomic DNA was isolated form a stationary culture of L21-Fru-ABT using the Jetflex Genomic DNA Purification Kit (GENOMED, Cat. no. 600100; Löhne, Germany) according to the protocol provided by the manufacturer with the modifications described previously (Ben Hania et al., 2015). The complete genome sequence was determined using a combination of two genomic libraries of which one was prepared for sequencing with the PacBio RSII (Pacific Biosciences, Menlo Park, CA, USA) and the other for the Illumina MiSeq platform (Illumina, San Diego, CA, USA). The SMRTbell template library was prepared and sequenced according to the instructions from Pacific Biosciences following the Procedure & Checklist ‘Greater than 10 kb Template Preparation and Sequencing’ applying C2 chemistry. In total, five SMRT cells were run, taking 120 min movies. Illumina sequencing was performed on a MiSeq platform with 2 × 150 cycles. The paired-end library contained inserts of an average insert size of 500 bp and delivered 1.8 million reads.
A draft long read genome assembly named L21-Fru-AB-SP4_HGAP_5SC_std_np was created using the ‘RS_HGAP_Assembly.1’ protocol included in SMRTPortal version 2.1.1 including all five SMRT cells applying parameters described previously (Ben Hania et al., 2015), but not allowing partial alignments. Finally, one chromosomal contig could be obtained, which was trimmed, circularized and adjusted to dnaA as first gene. A total coverage of 265 × has been calculated within the long read assembly process. DNA base modifications analysis was performed by ‘RS_Modification_and_Motif_Analysis.1’ protocol with default settings. Quality check of the final consensus sequences regarding overall coverage as well as SNPs was performed using IGV (Thorvaldsdóttir et al., 2013) after mapping of Illumina short read data onto the draft genome using BWA (Li and Durbin, 2009).
Genome annotation was primarily done using PROKKA version 1.8 (Seemann, 2014). The annotated genome was then compared with results provided by the RAST server (Overbeek et al., 2014). In cases where automatic annotation by RAST and PROKKA led to aberrant results, the function prediction of PROKKA was checked and eventually corrected manually by using BLASTP to search for similar proteins in the UniProtKB database (http://www.uniprot.org/blast/). Additional gene prediction analyses and functional annotation was performed within the Integrated Microbial Genomes—Expert Review (IMG-ER) platform (Markowitz et al., 2009). Genomic islands were localized with the SIGI-HMM program (Waack et al., 2006) of the IslandViewer 3 (Dhillon et al., 2015). The annotated complete genome sequence of L21-Fru-ABT was deposited in GenBank with the accession no. CP010904. The version described in this paper is the first version, CP010904.1.
Phylogenetic analyses
Comparative sequence analyses of almost complete 16S rRNA gene sequences or full-length protein sequences of the RNA polymerase beta subunit (RpoB) were used to determine the phylogenetic position of strain L21-Fru-ABT and related bacteria within the PVC superphylum. The 16S rRNA gene sequence of strain L21-Fru-ABT was determined previously (Spring et al., 2015) and deposited in the GenBank/EMBL/DDBJ databases under the accession number KC665948. The 16S rRNA sequence of L21-Fru-ABT was added to the alignment of the SILVA database (Quast et al., 2013; SSU Ref NR 99 release 119) using the integrated aligner of the ARB software package (Ludwig et al., 2004). On the basis of the curated guide tree included in the SSU Ref NR 99 database, a comprehensive data set of reference sequences representing major clades of the PVC superphylum were selected. Thereafter, remaining alignment errors revealed by visual inspection were corrected manually. Protein sequences were obtained from the GenBank or Uniprot databases and aligned using the MUSCLE algorithm (Edgar, 2004) implemented in the ARB package.
Phylogenetic trees based on aligned data sets of nucleotide or amino acid sequences were reconstructed using programs implemented in the ARB software package. When the ARB neighbor-joining program was used, phylogenetic distances were calculated with the corrections of Felsenstein or Jukes-Cantor for nucleotide sequences or with PAM for amino acid sequences. Maximum likelihood trees were reconstructed using RAxML (version 7.7.2) with the GTRGAMMA model for DNA and PROTCATLGF for protein data under the rapid bootstrap analysis algorithm. The robustness of the tree topologies was evaluated by performing 1000 rounds of bootstrap replicates.
Estimates of the phylogenetic diversity within the proposed Kiritimatiellaeota phylum and the related phyla Verrucomicrobia and Lentisphaerae were deduced applying the similarity option of the ARB distance matrix program. For the Kiritimatiellaeota phylum, a data set containing 16S rRNA gene sequences included in the guide tree of the SILVA 119 SSU Ref NR 99 data set and affiliated with subdivision 5, except sequences tagged as being of low quality were used. In a second more stringent calculation, sequences that were labeled as unclassified by two independent taxonomic frameworks (RDP and Greengenes) were also excluded from the data set. For the calculations of sequence identity values within the Verrucomicrobia and Lentisphaerae (except subdivision 5), all sequences assigned to these phyla in the SILVA 119 SSU Ref NR 99 data set were used.
The environmental distribution of members of the proposed phylum Kiritimatiellaeota was deduced from the source of isolation given for environmental clone sequences affiliated with subdivision 5 in the SILVA 119 SSU Ref NR 99 data set. Sequences with a different classification in taxonomic frameworks provided by RDP (Wang et al., 2007) or Greengenes (McDonald et al., 2012) were excluded from the analyses, because this may indicate an erroneous affiliation of these sequences to subdivision 5 in the SILVA database. A list of all used 16S rRNA gene sequences is presented in the Supplementary Table S1.
Determination of phenotypic traits and biochemical analyses
The shape and size of cells grown to stationary phase was observed with an AxioScope.A1 phase contrast microscope (Carl Zeiss, Jena, Germany) equipped with an AxioCam MRc digital camera controlled by the software Axio Vision version 4.8. Samples for electron microscopy were prepared according to the protocols described in Wittmann et al. (2014).
The cellular fatty acid pattern of strain L21-Fru-ABT was determined from cells grown at 28 °C to early stationary phase in basal medium containing 1.0 g l−1 d-glucose as carbon source. For comparison, cellular fatty acid patterns were obtained from related type strains that were cultured as indicated in the catalog of the Leibniz Institute DSMZ (see above). The preparation and extraction of fatty acid methyl esters from biomass and their subsequent separation and identification by gas chromatography was performed as described elsewhere (Miller, 1982; Kaksonen et al., 2006). Extraction and analyses of respiratory lipoquinones and polar lipids were carried out according to previously published protocols (Tindall, 1990; Tindall et al., 2007). Diagnostic diamino acids of the cell wall peptidoglycan were detected in hydrolysates of whole cells by using the gas chromatographic/mass spectrometric method described by Jeske et al. (2015). Samples of lyophilized biomass were supplemented with a defined amount of norleucine serving as internal standard for the quantification of diagnostic diamino acid derivatives on the basis of Total-Ion Current Chromatograms. The diamino acid derivatives were identified by their retention times approved by those of ornithine and 2,6-diaminopimelic acid derivatives obtained from authentic standards (Sigma O2250 and ALDRICH 33240, respectively) as well as by Extracted Ion Chromatograms based on characteristic mass spectrometric fragment ions.
The determination of growth parameters and physiological characteristics was performed according to the methods reported previously (Spring et al., 2013; Ben Hania et al., 2015). Non-gaseous fermentation products (alcohols, volatile and non-volatile fatty acids) were determined upon growth of strain L21-Fru-ABT in basal medium with 5 mM glucose as substrate using gas chromatography according to published protocols (Holdemann et al., 1977; Steer et al., 2001). Formate was quantified by a colorimetric assay (Lang and Lang, 1972).
Results and discussion
Source, enrichment and isolation of strain L21-Fru-ABT
Strain L21-Fru-ABT was obtained from a several centimetres thick photosynthetically active laminated microbial mat covering the bottom of the shallow hypersaline Lake 21 on the Kiritimati Atoll (Central Pacific). A unique characteristic of this cyanobacterial mat was a zone of disintegration observed at the transition between the photosynthetically active upper layer and a deep zone that remains permanently dark and anoxic. At this intermediate zone, the dense gelatinous mat turned into a flaked mucous layer that contained large evaporitic calcium sulfate deposits (gypsum aggregates). It was supposed that this transition zone represents the surface layer of an ancient microbial mat after being overgrown by the recent cyanobacterial mat (Spring et al., 2015). Anoxic samples of the Lake 21 microbial mat were used to inoculate various cultivation media for the enrichment of anaerobic saccharolytic bacteria that presumably have a major role in the observed disintegration process within the suboxic zone. From an enrichment culture containing fructose as substrate and supplemented with the antibiotic penicillin G to prevent growth of Gram-positive bacteria, strain L21-Fru-ABT could be isolated by applying the dilution-to-extinction technique. In support of a possible participation of this strain in the degradation of the mat matrix, 16S rRNA gene sequences with a high similarity to the sequence of L21-Fru-ABT were only retrieved from a distinct layer of the zone that was mainly affected by decay and disintegration (Spring et al., 2015).
Phylogenetic placement within the PVC superphylum and environmental distribution of related bacteria
The coherent cluster of environmental sequences comprising strain L21-Fru-ABT was originally defined as subdivision 5 of the Verrucomicrobia phylum by Hugenholtz et al. (1998). This traditionally used affiliation was recently questioned by the distributors of the ARB SILVA database release 119, who assigned sequences representing subdivision 5 to the Lentisphaerae phylum (Yilmaz et al., 2014, 2016). Owing to these conflicting results, a further detailed analysis of the phylogenetic placement of subdivision 5 within the PVC superphylum seems to be required.
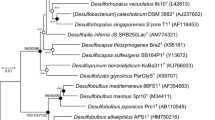
On the basis of the SILVA 119 SSU Ref NR 99 database, almost complete 16S rRNA gene sequences were selected that represent all recognized classes within the PVC superphylum and the major phylogenetic lineages of the subdivision 5. This data set was then used for thorough phylogenetic analyses applying maximum likelihood, maximum parsimony and distance algorithms. The representative 16S rRNA gene tree shown in Figure 1 confirms the placement of strain L21-Fru-ABT within the sequence cluster commonly referred to as subdivision 5. However, there is no recognizable association with the Lentisphaerae phylum thereby contradicting the assignment made in the SILVA 119 database. Despite a moderate bootstrap support for the affiliation of subdivision 5 with the Verrucomicrobia, a stable bifurcation has to be noted that separates this group from the other major clades representing the Verrucomicrobia phylum, which on the other hand cluster together with high bootstrap support. Furthermore, the apparent phylogenetic depth of subdivision 5 has a similar level compared with the divergence observed in the branch comprising the remaining clades of the Verrucomicrobia phylum. Thus, it is likely that the group of sequences previously recognized as the Verrucomicrobia subdivision 5 represents a separate phylum.

Phylogenetic placement of K. glycovorans L21-Fru-ABT within the PVC superphylum based on almost complete 16S rRNA gene sequences. The tree topology was reconstructed under the maximum likelihood criterion and rooted using the 16S rRNA gene sequence of Escherichia coli K12 (AKVX01000001, not shown). Names in quotation represent taxa that are not yet validly published. Support of a distinct branching by bootstrap analyses is indicated by symbols. Black dots at a distinct node indicate that bootstrap values of 95% or above (percentages of 1000 resamplings) were obtained with three different reconstruction methods, while gray dots indicate that values of 95% or above were obtained with only two reconstruction methods. White dots indicate that bootstrap values of 75% or above were obtained with at least one reconstruction method. In such cases, the values of 75% or above are given from left to right for the maximum likelihood, maximum parsimony and neighbor-joining method. Polygons represent clades of several sequences. Scale bar, 0.10 changes per nucleotide position.
Additional phylogenetic analyses based on a more comprehensive data set of 16S rRNA genes or RpoB proteins as alternative phylogenetic marker are presented in the Supplementary Material S1. In summary, it could be clearly shown that members of the subdivision 5 cannot be reliably associated with Verrucomicrobia or Lentisphaerae, but instead represent a sister lineage loosely associated with both phyla (Supplementary Figure S1). In a recent study by Yarza et al. (2014), taxonomic thresholds based on sequence identity values were suggested, which were deduced from an analysis of available 16S rRNA gene sequences of established taxa belonging to various ranks. For classes, the proposed thresholds for median and minimum sequence identity values were 86.35% and 80.38%, respectively, and accordingly for phyla 83.68% and 77.43%, respectively. The analysis of 915 high quality 16S rRNA gene sequences assigned to subdivision 5 in the SILVA 119 SSU Ref NR 99 data set revealed an average identity value of 82.3% and a minimum sequence identity of 69.4%. Even after restricting the data set to 836 sequences, which were correctly classified in RDP and/or Greengenes, the respective values were 82.7% and 72.4%, thereby confirming the taxonomic rank of a phylum. We determined for comparison also average and minimum sequence identities for related phyla and obtained values of 81.5% and 65.5%, respectively, for the Verrucomicrobia and 83.1% and 72.7%, respectively, for Lentisphaerae (except subdivision 5). Note that the minimum sequence identity value of 65.5% obtained for Verrucomicrobia may indicate that this phylum is currently not well defined, because the sequence identity value of sequences allocated to one phylum should be above 75.0% (Yarza et al., 2014).
The separate evolution of strain L21-Fru-ABT and members of the Verrucomicrobia phylum is also supported by an analysis of the phylogenetic distribution of genes in genomes of several representatives of the PVC superphylum. Figure 2 shows that the percentage of positive BLAST hits of assigned L21-Fru-ABT proteins with protein-coding genes of members of the Verrucomicrobia is below 10% and thus in the range obtained with members of other phyla, whereas strains representing the established five main lineages of Verrucomicrobia displayed values of 40% or above. Consequently, it seems justified to establish a novel phylum for members of the so far only tentatively defined subdivision 5, which is named Kiritimatiellaeota referring to the geographical origin of the first cultured strain, the atoll of Kiritimati in the Central Pacific.

Phylogenetic composition of the genomes of K. glycovorans L21-Fru-ABT and several representative members of the PVC superphylum. The bar chart is based on original data retrieved form the IMG/ER server and displays percentages of protein-coding genes with a positive BLAST hit in genomes of a distinct phylum. The threshold was set to 30% or more sequence identity. Unassigned proteins were not included in the calculation of the phylogenetic distribution. Separate values are only shown for phyla that had 10% or more hits with at least one sample. The following strains and genome sequences were used (IMG Genome IDs in parentheses): Verrucomicrobium spinosum DSM 4136T (641736179); ‘Chthoniobacter flavus’ Ellin428 (642791618); ‘Pedosphaera parvula’ Ellin514 (645058870); ‘Ca. Methylacidiphilum infernorum’ V4 (642555138); Opitutus terrae PB90-1T (641522643); K. glycovorans L21-Fru-ABT (2606217195); Lentisphaera araneosa HTCC2155T (640963040); Planctopirus limnophila DSM 3776T (646564559).
The evolutionary divergence between members of the Verrucomicrobia and the proposed Kiritimatiellaeota phylum is also reflected in a different habitat preference, thereby confirming the postulated ecological coherence of high taxonomic ranks (Philippot et al., 2010). The majority of representatives of the subdivisions 1, 2, 3, 4 and 6 of the Verrucomicrobia phylum inhabit soil, freshwater or marine environments and are expected to have a metabolism adapted to aerobic or microaerophilic conditions (Hedlund, 2011). In contrast, as shown in Figure 3, members of the Kiritimatiellaeota phylum occupy predominantly niches characterized by anoxic conditions, like the intestine of animals, wastewater or hypersaline sediments, whereas only few representatives were detected in soil or the water column of marine and freshwater habitats. Strain L21-Fru-ABT could be assigned to the R76-B128 clade, which occurs mainly in hypersaline and marine sediments. A further detailed analysis of the environmental distribution of members of distinct clades of the proposed Kiritimatiellaeota phylum is presented in the Supplementary Material S1.

Environmental distribution and habitat-specific phylogenetic structure of the proposed Kiritimatiellaeota phylum. (a) Pie chart diagram illustrating the relative proportions of environmental sources of 913 distinct 16S rRNA clone sequences according to the SILVA 119 SSU Ref NR 99 database. A list of the used 16S rRNA gene sequences is available in Supplementary Table S1. (b) Phylogenetic tree of the same set of sequences reconstructed with the ARB neighbor-joining program using the sequence of Verrucomicrobium spinosum DSM 4136T as outgroup (NR_026266, not shown). Phylogenetic distances were calculated with the correction of Felsenstein as implemented in the ARB neighbor-joining program. Sequences of distinct clades are collapsed to polygons and the number of sequences comprising a clade is shown within the corresponding polygon. The environmental distribution of sequences assigned to a distinct clade is shown as pie chart next to the respective polygon. The prevailing source of the sequences is reflected by the colors of the polygons. Scale bar, 10% estimated sequence divergence.
General features of the genome
The genome of strain L21-Fru-ABT is represented by one circular chromosome that has a total length of 2 949 723 bp and a G+C content of 63.31 mol%. High proportions of genes assigned to COG functional categories are involved in the transport and metabolism of carbohydrates (129) and amino acids (128), while the metabolism of nucleotides (54) and lipids (52) has a minor role. A general preference for the metabolism of carbohydrates appears to be a typical characteristic of free-living and chemoheterotrophic members of the PVC superphylum, which are often specialized on the degradation of recalcitrant polysaccharides and glycopolymers (for example, Glöckner et al., 2003; Martinez-Garcia et al., 2012). The genome is further characterized by a prevalence of transposase genes (75) and tentative genomic islands (12), which could reflect an increased flexibility of the genome structure caused by a recent genetic isolation of this strain following the adaptation to hypersaline mats. A further detailed analysis of the genome sequence is presented in the Supplementary Material S2. In addition, a list of manually annotated genes used to deduce molecular or phenotypic traits of strain L21-Fru-ABT is available in the Supplementary Table S2.
Morphological and biochemical traits
Cells of strain L21-Fru-ABT had a regular spherical shape, a size of 1–2 μm, were non-motile and divided by binary fission; sometimes, larger cells of up to 3 μm diameter were observed. No colonies were formed on agar plates incubated in anaerobic jars or under aerobic conditions, which could explain why similar strains were not isolated earlier by the frequently used plating of environmental samples. In liquid medium, aggregates comprising several hundreds of cells were formed at the beginning of the growth phase or under conditions of nutrient limitation (Figure 4a). In most cases, the center of aggregates consisted of diffuse material, most likely extracellular polymeric substances, to which the cells were attached. The production of exopolymers is also indicated by scanning electron micrographs of liquid culture samples, which show the interconnection of cells by a fibrillary network that became apparent upon fixation and dehydration of the sample (Figure 4b). The formation of spores was not observed and no sporulation genes were annotated in the genome sequence. Cellular reserve materials stored in granules were not directly observed or identified using microscopic methods, but based on the annotated genome sequence, it can be deduced that in this strain, inorganic polyphosphates and glycogen are stored as carbon and energy reserve (see Supplementary Material S2). In stained thin sections of cells, often distinct electron-dense regions became apparent by transmission electron microscopy (Figure 4c), which could indicate a DNA-containing nucleoid. No further compartmentalization of the cytoplasm or intracytoplasmic membrane invaginations could be detected. Cells stained Gram-negative and cell walls visualized by electron microscopy had a structure typical of Gram-negative bacteria comprising an outer membrane, a periplasmic space and a cytoplasmic membrane (Figure 4d). A canonical Gram-negative cell plan is also supported by a large number of genes encoding putative enzymes involved in peptidoglycan and lipopolysaccharide synthesis as well as a cell division complex comprising FtsZ (see Supplementary Material S2).

Shape and ultrastructure of cells of K. glycovorans L21-Fru-ABT. (a) Phase contrast micrograph of an aggregate formed by cells upon cultivation in liquid medium. (b) Scanning electron micrograph of a fixed sample of cells grown in liquid medium showing the formation of extracellular polymeric substances. (c) Transmission electron micrograph of a negatively stained thin section showing a condensed dark region probably representing the nucleoid. (d) Enlarged view of the rectangular area marked in (c). Arrows mark characteristic features of a Gram-negative cell envelope: outer membrane (om), periplasmic space (ps) and cytoplasmic membrane (cm).
A gas chromatographic/mass spectrometric analysis of whole-cell hydrolysates of strain L21-Fru-ABT revealed the presence of ornithine (Orn) and 2,6-diaminopimelic acid (Dap) as diagnostic diamino acids of peptidoglycan. Interestingly, Orn could be also identified in whole-cell hydrolysates of Victivallis vadensis and Oligosphaera ethanolica, two representatives of the Lentisphaerae phylum (Supplementary Material S3). It is noteworthy that this would represent the first report about the detection of peptidoglycan with Orn as the diagnostic diamino acid in bacteria with a Gram-negative type of cell wall that do not belong to the Spirochaetaceae. However, it cannot be excluded that Orn could have been extracted from certain aminolipids or other cell components instead of peptidoglycan, because only whole-cell hydrolysates were analyzed. Further details about the chemotaxonomic characterization of strain L21-Fru-ABT including its polar lipid and cellular fatty acid composition are presented in the Supplementary Material S3 and the formal species description at the end of this paper.
The genome of strain L21-Fru-ABT contained no genes involved in the production of respiratory lipoquinones, cytochromes or terminal oxidases. Accordingly, biochemical tests for the presence of respiratory lipoquinones, oxidase and catalase were negative. The absence of elements of a membrane-bound electron transport chain indicates a strictly anaerobic and fermentative type of metabolism.
Physiology of growth
Determination of the optimal growth conditions characterized strain L21-Fru-ABT as a mesophilic, neutrophilic and moderate halophilic bacterium thereby reflecting an adaptation to hypersaline lakes, the environment from which it was isolated (Table 1). Under optimal growth conditions in complex medium, the mean generation time was 22 h. Strain L21-Fru-ABT was quite aerotolerant and could proliferate in the presence of oxygen while reducing the medium, which may be an important trait for growth in cyanobacterial mats known to produce excess oxygen by daylight. However, growth in the presence of oxygen was not stable and ceased after the second transfer in aerobic medium, which was probably because of the accumulation of oxidative damage caused by reactive oxygen species or the inhibition of oxygen-sensitive enzymes involved in central metabolic pathways.
On the basis of the annotated genome sequence, it can be predicted that in strain L21-Fru-ABT, glycine betaine and sucrose represent the main osmoprotectants to balance changes of extracellular salt concentrations (see Supplementary Material S2). The reserve polymer glycogen could be used to keep glucose available as precursor for sucrose synthesis. Interestingly, it has been assumed originally that cyanobacteria and proteobacteria represent the only prokaryotes that produce sucrose as compatible solute, but with the increase of available genome sequences, genes involved in the production of sucrose were also detected in members of the phyla Deferribacteres, Chrysiogenetes, Firmicutes and Ignavibacteriae (Diricks et al., 2015). In this study, genes of the key enzymes sucrose synthase and sucrose-6 F-phosphate phosphohydrolase were detected for the first time in a member of the PVC superphylum.
The determined substrate utilization spectrum of strain L21-Fru-ABT was quite narrow and comprised mainly monosaccharides, whereas amino acids, carboxylic acids or alcohols were not utilized. The main non-gaseous end products of glucose fermentation were identified as ethanol (1–2 mM), acetate (<1 mM) and lactate (traces). The total amount of produced fermentation products was quite low and indicates that only a small amount of glucose was metabolized during growth. Production of biomass and fermentation products could not be increased by using higher concentrations of glucose (above 5 mM), which indicates a metabolism adapted to oligotrophic growth conditions. The gas that accumulated in the headspace of cultures during growth of this strain was most likely hydrogen, which may be produced by two distinct bifurcating cytoplasmic iron-only hydrogenases encoded at adjacent sites of the genome (Supplementary Table S2).
Despite the low number of utilized carbon sources, the genome of strain L21-Fru-ABT encodes an array of 66 glycoside hydrolases and 56 sulfatases, which may indicate that various sulfated glycopolymers are utilized as food source by this strain. An alternative function of sulfatases would represent the utilization of sulfated compounds as sources of sulfur as described by Kertesz (2000). However, the numbers of genes annotated as sulfatases in genomes of members of the phylum Verrucomicrobia show in general a positive correlation with the salinity of the preferred habitat and thus tend to be higher in environments characterized by large amounts of free sulfate (Figure 5). This finding is in line with the general belief that sulfatases are required for the degradation and subsequent utilization of sulfated glycopolymers as carbon source in marine strains of Planctomycetes (Glöckner et al., 2003; Wegner et al., 2013). Compared with marine strains of the phylum Verrucomicrobia, the genome sequence of strain L21-Fru-ABT revealed an expansion of sulfatase genes in proportion to the genome size, which could indicate a specialization on the utilization of sulfated exopolymers. Although the tested commercially available sulfated glycopolymers did not allow stable growth of this strain under laboratory conditions, a plethora of alternative sulfated compounds can be found in hypersaline microbial mats and may represent suitable substrates for growth.

Habitat dependence of the prevalence of sulfatase genes in the genomes of K. glycovorans L21-Fru-ABT and other saccharolytic strains belonging to the Verrucomicrobia. For each strain, the number of sulfatase genes and their frequency in the respective genome is shown. The preferred habitats of the analyzed strains are symbolized by the colors of the dots. The size of the dots correlates with the genome size in mega base pairs (Mb). Broken lines mark distinct clusters of strains preferring the same environment.
It is well known, for example, that halophilic cyanobacteria produce sulfated exopolysaccharides (De Philippis et al., 1998) and are able to replace membrane-bound phospholipids with sulfoglycolipids like sulfoquinovosyldiacylglycerol, especially under conditions of phosphate limitation (Van Mooy et al., 2009). Furthermore, sulfated glycoproteins that may represent an essential component of the extracellular polymeric substance matrix in distinct zones of the mat could represent a suitable nutrient source. Interestingly, genes of several hydrolytic enzymes that have been associated with the degradation of O-linked oligosaccharides of the sulfated glycoprotein mucin (Tailford et al., 2015) were detected in the genome of strain L21-Fru-ABT, including neuraminidase (L21SP4_01677), α-l-fucosidase (L21SP4_01318), and a combined sulfatase and α-N-acetylglucosaminidase (L21SP4_01423). A list of all glycoside hydrolase families identified in strain L21-Fru-ABT by the CAZy database (Lombard et al., 2014) is shown in Supplementary Table S3. Members of families GH78 and GH106, which are mainly active as α-l-rhamnosidases, seem to be dominating in this strain. This could indicate that bioactive compounds containing l-rhamnose represent the main target for degradation. Several studies have demonstrated that exopolymers containing rhamnose have an important function for the establishment and maintenance of bacterial biofilms (for example, Davey et al., 2003; Michael et al., 2016). Consequently, a specific degradation of such glycopolymers by strain L21-Fru-ABT could contribute to the disintegration of the mat matrix observed in the decaying zone of the mat. In addition, rhamnose has been identified as a main component of the sulfated polysaccharide mucilage secreted by marine diatoms (Passow, 2002). However, stable growth with l-rhamnose as substrate could not be demonstrated in laboratory experiments, although all necessary genes for its uptake and catabolism were present in the genome (Supplementary Table S2). In general, it was observed that growth of this strain with certain carbon sources (for example, galactose, fucoidan, carrageenan, mucin) was unstable and ceased upon the second transfer, which could indicate that the enzymes required for the utilization of these substrates were expressed only transiently. An alternative explanation would be that in the natural environment, a consortium of diverse strains participates in the breakdown of complex recalcitrant compounds and strain L21-Fru-ABT is specialized only on a few distinct steps of the degradation process, which leads to nutrient limitation in axenic culture.
More than 50% of the glycoside hydrolases identified by the CAZy database and 35% of the predicted sulfatases are tagged with signal peptides that guide their integration into a cell membrane or export into the periplasmic or extracellular space, where they could degrade large or insoluble biopolymers that cannot be transported into the cell. On the basis of the genome sequence, several transport systems for the membrane translocation and secretion of proteins could be identified, including the Sec preprotein-translocation pathway, the twin-arginine translocation mechanism and a type II secretion system (see Supplementary Material S2). It is known that polymer-degrading members of the Bacteroidetes use TonB-dependent receptors for the transport of partially degraded polysaccharides into the periplasm (Kabisch et al., 2014), probably to avoid exploitation of released sugar monomers by microbial competitors (‘cheaters’). However, no genes encoding such uptake systems were detected in strain L21-Fru-ABT.
Core metabolism and regulation
The obtained genome sequence of strain L21-Fru-ABT allowed for the first time a reconstruction of the central metabolism of a member of a phylum-level lineage of bacteria previously recognized as Verrucomicrobia subdivision 5. Routes of main metabolic pathways can be proposed based on the annotation of genes listed in the Supplementary Table S2. All enzymes for the oxidation of glucose to pyruvate by the Embden–Meyerhof–Parnas pathway (glycolysis) were present. However, no gene for pyruvate kinase that catalyzes the irreversible formation of pyruvate and ATP from phosphoenolpyruvate and ADP could be detected, although it was present in all other available genomes of members of the phyla Verrucomicrobia and Lentisphaerae. Instead, in L21-Fru-ABT, the enzyme pyruvate phosphate dikinase (L21SP4_01945) is probably used and catalyzes a reversible reaction that depends on AMP and diphosphate for the synthesis of ATP from phosphoenolpyruvate. Interestingly, it has been previously reported that in some anaerobic unicellular eukaryotes and Clostridium symbiosum, pyruvate phosphate dikinase catalyzes the terminal step of glycolysis and functions primarily in the direction of ATP synthesis (Reeves, 1971). The advantage of using pyruvate phosphate dikinase instead of pyruvate kinase is an increase of the ATP yield of glycolysis, especially in combination with the enzymes pyrophosphate-fructose 6-phosphate 1-phosphotransferase and adenylate kinase (Chastain et al., 2011), which are both encoded in the genome of L21-Fru-ABT and make use of the energy-rich compound pyrophosphate. As illustrated in the pathway diagram in Supplementary Material S2, this atypical and probably ancient glycolytic pathway yields five instead of two mol ATP per mol glucose and may be therefore of special value in anaerobic, energy-limited environments.
In addition to substrate-level phosphorylation, strain L21-Fru-ABT is probably capable of using a chemiosmotic mechanism for the production of ATP. An electrochemical gradient may be generated for example by a membrane-bound RNF complex and then used by a proton- or Na+-translocating V-type ATP-synthase (see Supplementary Material S2). Prediction of the preferred ion for building up an electrochemical gradient is still difficult by means of bioinformatic analyses, but based on the environmental growth conditions of this strain (anoxic and hypersaline), we speculate that Na+ may be used as coupling ion for ATP synthesis, because in such habitats, sodium gradients have some advantages against using a proton-motive force (Mulkidjanian et al., 2008; Spring et al., 2010).
Strain L21-Fru-ABT is able to grow in mineral medium with glucose as the sole substrate and therefore should have the genetic inventory to produce all necessary enzymes for the biosynthesis of its cellular compounds. Interestingly, isopentenyl pyrophosphate as precursor for isoprenoid synthesis is produced in strain L21-Fru-ABT via the mevalonate pathway with acetyl-CoA as starting material. Apart from the obligate intracellular chlamydial species Waddlia chondrophila (Bertelli et al., 2010), this strain represents so far only the second known member of the PVC phylum, which seems to use this pathway. All other genome-sequenced representatives of the phyla Verrucomicrobia, Lentisphaerae and Planctomycetes have the methylerythritol pathway for isoprenoid synthesis. Phylogenetic analyses of enzymes of the mevalonate pathway in members of the PVC superphylum are shown in the Supplementary Material S2. Interestingly, it turned out that the enzymes of strain L21-Fru-ABT and Waddlia chondrophila do not share the same evolutionary history, but can be affiliated with proteins of Proteobacteria and Spirochaetes, respectively. Therefore, a potential influence of lateral gene transfer events in the evolution of the mevalonate pathway in these species cannot be excluded.
The regulation of central metabolic pathways in strain L21-Fru-ABT is possibly controlled by a phosphorylation cascade that resembles a rudimentary phosphotransferase system for the group translocation of sugars (see Supplementary Material S2). In the proposed global regulatory network, the central metabolites phosphoenolpyruvate and pyruvate could be used to sense the availability of a suitable carbon source and phosphate. The inferred metabolic state of the cell would then be signaled to proteins controlling important cellular processes like the homeostasis of pH and sodium.
On the basis of the results of the genome analysis of strain L21-Fru-ABT, a metabolic model is proposed and presented in Figure 6.

Overview of the proposed central metabolism of K. glycovorans L21-Fru-ABT. The shown cartoon represents a summary of the information deduced from analysis of the genome sequence and is based on the manual annotated genes listed in Supplementary Table S2. Functional proteins are grouped in four sectors according to their role in metabolism. Distinct enzymes are distinguished by different colors that correspond with their assumed functions. The color code for the respective sectors is as follows. Nutrient uptake: turquoise, permease; red, ABC-type transporter; purple, NTP-dependent permease; yellow, outer membrane receptor; orange, outer membrane transport energization complex; green, TRAP-type C4-dicarboxylate transporter. Secretion and resistance: orange, twin-arginine translocation (TAT) complex; yellow, type II secretion system; red, Sec translocation complex; green, Wzx/Wzy-dependent biopolymer export complex; purple, TolC-dependent multidrug-resistance efflux pump; turquoise, permease. Stress response: green, NADH-quinone reductase; red, ABC-type transporter; turquoise, permease; purple, cation:proton antiporter. Central metabolism and regulation: red, V-type ATP-synthase; green, RNF complex; purple, membrane-bound adenylate-cyclase; turquoise, iron-only bifurcating hydrogenase. R-Fe3+, ferric iron siderophore complex; DC, dicarboxylic acid; NAG, N-acetylglucosamine; AA, amino acid; DPG, 1,3-diphosphoglycerate; PEP, phosphoenolpyruvate; Fd, ferredoxin.
Concluding remarks and classification
Characterization of the first cultured representative of the Verrucomicrobia subdivision 5 unveiled the lifestyle of an anaerobic saccharolytic bacterium that thrives in the suboxic zone of photosynthetically active laminated mats. Specific adaptations of strain L21-Fru-ABT to hypersaline cyanobacterial mats include a high oxygen and salinity tolerance as well as a diversified genomic inventory of glycoside hydrolases and sulfatases required for the degradation of recalcitrant sulfated glycopolymers. A general preference of members of subdivision 5 for anoxic environments at liquid–solid interfaces, which are often characterized by the formation of biofilms (for example, saline sediments) or mucous layers (animal intestine), could indicate that foraging on sulfated glycopolymers produced in such habitats is a key metabolic feature preserved in most members of this evolutionary lineage. In this regard, it is noteworthy that so far, members of subdivision 5 were not detected in the feces of humans or mice despite their prevalence in the intestine of other vertebrates, like for instance wild gorillas (Frey et al., 2006) or horses (Costa et al., 2015). It is tempting to speculate that this distribution pattern could be the result of a competition with other gut bacteria for the same ecological niche. If this is true, members of subdivision 5 could have been displaced in the colon of humans and mice by mucin-degrading Akkermansia species, which are efficiently colonizing the large intestine of both hosts (Donaldson et al., 2016).
On the basis of its determined phylogenetic position, strain L21-Fru-ABT represents a novel species and genus affiliated with a phylum-level lineage previously recognized as Verrucomicrobia subdivision 5. The results of the phylogenetic analyses were supported by comparative genome analyses, which revealed several distinguishing features of strain L21-Fru-ABT that were not present in the available genomes of bacteria representing neighboring phyla. Further differential phenotypic traits of strain L21-Fru-ABT to representatives of related phyla are summarized in Table 1. Formal descriptions of the proposed novel species and taxa of higher ranks follow below.
Description of Kiritimatiella gen. nov.
Kiritimatiella (Ki.ri.ti.ma.ti.el'la. N.L. fem. dim. n. Kiritimatiella, pertaining to the Kiritimati Atoll, the geographical origin of the type strain of the type species).
Free-living, non-motile and non-sporeforming coccoid cells that divide by binary fission. Intracellular membranes are not formed. Gram-negative type of cell wall with a peptidoglycan layer. Major cellular fatty acids are iso-C14:0 and C18:0. Odd-numbered cellular fatty acids, respiratory lipoquinones or cytochromes are not formed. Tests for catalase and oxidase are negative. Nitrate is not reduced. Obligately anaerobic, moderately halophilic, mesophilic and neutrophilic. Strictly fermentative metabolism with sugars as preferred substrates. Yeast extract or vitamins are not required for growth.
The type species is Kiritimatiella glycovorans.
Description of Kiritimatiella glycovorans sp. nov.
Kiritimatiella glycovorans (gly.co.vo'rans. N.L. pref. glyco- from Gr. adj. glukus sweet, referring to glucose; L. part. adj. vorans eating, devouring; N.L. part. adj. glycovorans eating glucose) shows the following characteristics in addition to those given for the genus. Most cells have a diameter of 1–2 μm; sometimes formation of aggregates with a size of up to 100 μm can be observed. Optimal conditions for growth are 28 °C, pH 7.5 and a salinity of 60–70 g l−1. The following substrates support stable growth: d-glucose, N-acetylglucosamine, d-mannose and d-xylose. The following compounds were tested, but not utilized under laboratory conditions: Tryptone, Casamino acids, yeast extract, mucin; l-alanine, l-isoleucine, l-glutamic acid; acetate, acrylate, benzoate, butyrate, citrate, fumarate, dl-lactate, dl-malate, 3-phenylpropionate, propionate, pyruvate, succinate; butanol, ethanol, glycerol, d-mannitol, methanol, propanol; sodium alginate, carrageenan, chitin, chondroitin sulfate, dextran (M.W. 170000), fucoidan, pullulan, starch, xanthan gum; l-arabinose, d-cellobiose, d-fructose, l-fucose, d-galactose, lactose, d-maltose, d-melibiose, l-rhamnose, d-ribose, sucrose and trehalose. The non-gaseous end products resulting from d-glucose fermentation are ethanol, acetate and lactate (traces). Ammonium was the only used nitrogen source, while nitrate, nitrite, urea, l-glutamic acid or molecular nitrogen were not utilized. The polar lipid composition is characterized by high amounts of phosphatidylglycerol, minor amounts of an unidentified phospholipid, two distinct glycolipids, an aminolipid and small amounts of phosphoglycolipids. In addition to the major fatty acids listed in the description of the genus, significant amounts of iso-C18:0, C12:0, C16:0 3OH, C20:0 and C16:0 are present, while anteiso-C15:0 is not detected. Susceptible to the antibiotics d-cycloserine, chloramphenicol A and tetracycline (each at 10 mg l−1). Ampicillin, carbenicillin, penicillin G and gentamicin are tolerated at least in concentrations up to 100 mg l−1, while kanamycin A is tolerated up to 1000 mg l−1. The DNA G+C content of the type strain is 63.3 mol%.
The type strain is L21-Fru-ABT (=DSM 26986T=JCM 19195T), isolated from the suboxic zone of a hypersaline microbial mat at the littoral zone of Lake 21, Kiritimati, Republic of Kiribati.
Description of Kiritimatiellaceae fam. nov.
Kiritimatiellaceae (Ki.ri.ti.ma.ti.el.la.ce'ae. N.L. fem. dim. n. Kiritimatiella, type genus of the family; suff. -aceae, ending to denote a family; N.L. fem. pl. n. Kiritimatiellaceae, the Kiritimatiella family).
The family encompasses mainly bacteria found in hypersaline and anoxic environments. Cells divide by binary fission, do not form intracellular membranes and have a cell wall of the Gram-negative type that contains peptidoglycan.
The affiliation of novel species to this family depends on the phylogenetic position, which should be determined on the basis of comparative sequence analyses of 16S rRNA and/or rpoB genes. The 16S rRNA sequence identity values of newly described strains affiliated with this family should be above 86.5% to the type strain of the type species Kiritimatiella glycovorans, which represents the threshold recommended by Yarza et al. (2014) for the definition of families.
The type genus of the family is Kiritimatiella.
Description of Kiritimatiellales ord. nov.
Kiritimatiellales (Ki.ri.ti.ma.ti.el.la'les. N.L. fem. dim. n. Kiritimatiella, type genus of the order; suff. -ales, ending to denote an order; N.L. fem. pl. n. Kiritimatiellales, the Kiritimatiella order).
Cells divide by binary fission, do not form intracellular membranes and have a cell wall of the Gram-negative type that contains peptidoglycan.
The affiliation of novel species to this order depends on the phylogenetic position, which should be determined on the basis of comparative sequence analyses of 16S rRNA and rpoB genes. The 16S rRNA sequence identity values of newly described strains affiliated with this order should be above 82.0% to the type strain of the type species Kiritimatiella glycovorans, which represents the threshold recommended by Yarza et al. (2014) for the definition of orders.
The type genus of the order is Kiritimatiella.
Description of Kiritimatiellae classis nov.
Kiritimatiellae (Ki.ri.ti.ma.ti.el'lae. N.L. fem. dim. n. Kiritimatiella, type genus of the type order of the class; N.L. fem. pl. n. Kiritimatiellae, the class of the order Kiritimatiellales).
The description is the same as for the order, except that a 16S rRNA gene sequence identity value of 78.5% to the type strain of the type species Kiritimatiella glycovorans should be used as threshold for the affiliation of strains to this class.
The type order of the class is Kiritimatiellales.
Description of Kiritimatiellaeota phyl. nov.
Kiritimatiellaeota (Ki.ri.ti.ma.ti.el'lae'ota. N.L. fem. dim. n. Ki.ri.ti.ma.ti.el'lae, type class of the phylum; suff. -aeota, ending to denote a phylum; N.L. fem. pl. n. Kiritimatiellaeota, the phylum of the class Kiritimatiellae).
Members of this phylum form a stable lineage that is separate from the Verrucomicrobia and Lentisphaerae phyla. The group of phylotypes representing this phylum and the encountered phylogenetic depth corresponds to the Verrucomicrobia subdivision 5 as originally defined by Hugenholtz et al. (1998). The main phenotypic characteristics are as given for the order.
The type class of the phylum is Kiritimatiellae.
References
Ben Hania W, Joseph M, Schumann P, Bunk B, Fiebig A, Spröer C et al. (2015). Complete genome sequence and description of Salinispira pacifica gen. nov., sp. nov., a novel spirochaete isolated form a hypersaline microbial mat. Stand Genomic Sci 10: 7.
Bergmann GT, Bates ST, Eilers KG, Lauber CL, Caporaso JG, Walters WA et al. (2011). The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol Biochem 43: 1450–1455.
Bertelli C, Collyn F, Croxatto A, Rückert C, Polkinghorne A, Kebbi-Beghdadi C et al. (2010). The Waddlia genome: a window into chlamydial biology. PLoS One 5: e10890.
Bryant M . (1972). Commentary on the Hungate technique for culture of anaerobic bacteria. Am J Clin Nutr 25: 1324–1328.
Cardman Z, Arnosti C, Durbin A, Ziervogel K, Cox C, Steen AD et al. (2014). Verrucomicrobia are candidates for polysaccharide-degrading bacterioplankton in an Arctic fjord of Svalbard. Appl Environ Microbiol 80: 3749–3756.
Chastain CJ, Failing CJ, Manandhar L, Zimmerman MA, Lakner MM, Nguyen THT . (2011). Functional evolution of C4 pyruvate, orthophosphate dikinase. J Exp Bot 62: 3083–3091.
Chin KJ, Liesack W, Janssen PH . (2001). Opitutus terrae gen. nov., sp. nov., to accommodate novel strains of the division ‘Verrucomicrobia’ isolated from rice paddy soil. Int J Syst Evol Microbiol 51: 1965–1968.
Costa MC, Silva G, Ramos RV, Staempfli HR, Arroyo LG, Kim P et al. (2015). Characterization and comparison of the bacterial microbiota in different gastrointestinal tract compartments in horses. Vet J 205: 74–80.
Davey ME, Caiazza NC, O’Toole GA . (2003). Rhamnolipid surfactant production affects biofilm architecture in Pseudomonas aeruginosa PAO1. J Bacteriol 185: 1027–1036.
De Philippis R, Margheri MC, Materassi R, Vincenzini M . (1998). Potential of unicellular cyanobacteria from saline environments as exopolysaccharide producers. Appl Environ Microbiol 64: 1130–1132.
Derrien M, Vaughan EE, Plugge CM, de Vos WM . (2004). Akkermansia municiphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol 54: 1469–1476.
Dhillon BK, Laird MR, Shay JA, Winsor GL, Lo R, Nizam F et al. (2015). IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res 43: W104–W108.
Diricks M, De Bruyn F, Van Daele P, Walmagh M, Desmet T . (2015). Identification of sucrose synthase in nonphotosynthetic bacteria and characterization of the recombinant enzymes. Appl Microbiol Biotechnol 99: 8465–8474.
Donaldson GP, Lee SM, Mazmanian SK . (2016). Gut biogeography of the bacterial microbiota. Nat Rev Microbiol 14: 20–32.
Edgar RC . (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797.
Fieseler L, Horn M, Wagner M, Hentschel U . (2004). Discovery of the novel candidate phylum ‘Poribacteria’ in marine sponges. Appl Environ Microbiol 70: 3724–3732.
Frey JC, Rothman JM, Pell AN, Nizeyi JB, Cranfield MR, Angert ER . (2006). Fecal bacterial diversity in a wild gorilla. Appl Environ Microbiol 72: 3788–3792.
Glöckner FO, Kube M, Bauer M, Teeling H, Lombardot T, Ludwig W et al. (2003). Complete genome sequence of the marine planctomycete Pirellula sp. strain 1. Proc Natl Acad Sci USA 100: 8298–8303.
Gupta RS, Bhandari V, Naushad HS . (2012). Molecular signatures for the PVC clade (Planctomycetes, Verrucomicrobia, Chlamydiae, and Lentisphaerae of bacteria provide insights into their evolutionary relationships. Front Microbiol 3: 1–19.
Hedlund BP. (2011). Phylum XXIII Verrucomicrobia phyl nov. In: Krieg NR, Staley JT, Hedlund BP, Paster BJ, Ward N, Ludwig W et al. (eds), Bergey’s Manual of Systematic Bacteriology, Vol 4. Springer Verlag: New York, NY, USA, pp 795–841.
Holdemann LV, Cato EP, Moore WEC . (1977) Anaerobe Laboratory Manual, 4th edn. Anaerobe Laboratory, Virginia Polytechnic Institute and State University: Blacksburg, VA, USA.
Horn M, Collingro A, Schmitz-Esser S, Beier CL, Purkhold U, Fartmann B et al. (2004). Illuminating the evolutionary history of chlamydiae. Science 304: 728–730.
Hugenholtz P, Goebel BM, Pace NR . (1998). Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180: 4765–4774.
Hungate R . (1950). The anaerobic mesophilic cellulolytic bacteria. Bacteriol Rev 14: 1–49.
Jeske O, Schüler M, Schumann P, Schneider A, Boedeker C, Jogler M et al. (2015). Planctomycetes do possess a peptidoglycan cell wall. Nat Commun 6: 7116.
Jetten MS, Wagner M, Fuerst J, Van Loosdrecht M, Kuenen G, Strous M . (2001). Microbiology and application of the anaerobic ammonium oxidation (’anammox') process. Curr Opin Biotechnol 12: 283–288.
Kabisch A, Otto A, König S, Becher D, Albrecht D, Schüler M et al. (2014). Functional characterization of polysaccharide utilization loci in the marine Bacteroidetes ‘Gramella forsetii’ KT0803. ISME J 8: 1492–1502.
Kaksonen AH, Spring S, Schumann P, Kroppenstedt RM, Puhakka JA . (2006). Desulfotomaculum thermosubterraneum sp. nov., a thermophilic sulfate-reducer isolated from an underground mine located in a geothermally active area. Int J Syst Evol Microbiol 56: 2603–2608.
Kamke J, Rinke C, Schwientek P, Mavromatis K, Ivanova N, Sczyrba A et al. (2014). The candidate phylum Poribacteria by single-cell genomics: New insights into phylogeny, cell-compartmentation, eukaryote-like repeat proteins, and other genomic features. PLoS One 9: e87353.
Kertesz MA . (2000). Riding the sulfur cycle—metabolism of sulfonates and sulfate esters in gram-negative bacteria. FEMS Microbiol Rev 24: 135–175.
Lang E, Lang H . (1972). Spezifische Farbreaktion zum direkten Nachweis der Ameisensäure. Fresenius’ Zeitschrift für Anal Chemie 260: 8–10.
Li H, Durbin R . (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760.
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B . (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42: 490–495.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC . (2009). IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 25: 2271–2278.
Martinez-Garcia M, Brazel DM, Swan BK, Arnosti C, Chain PSG, Reitenga KG et al. (2012). Capturing single cell genomes of active polysaccharide degraders: an unexpected contribution of Verrucomicrobia. PLoS One 7: e35314.
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6: 610–618.
Michael V, Frank O, Bartling P, Scheuner C, Göker M, Brinkmann H et al. Biofilm plasmids with a rhamnose operon are widely distributed determinants of the ‘swim-or-stick’ lifestyle in roseobacters. ISME J e-pub ahead of print 8 March 2016.
Miller LT . (1982). Single derivatization method for routine analysis of bacterial whole-cell fatty acid methyl esters, including hydroxy acids. J Clin Microbiol 16: 584–586.
Mulkidjanian AY, Galperin MY, Makarova KS, Wolf YI, Koonin EV . (2008). Evolutionary primacy of sodium bioenergetics. Biol Direct 3: 13.
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42: D206–D214.
Passow U . (2002). Transparent exopolymer particles (TEP) in aquatic environments. Prog Oceanogr 55: 287–333.
Philippot L, Andersson SG, Battin TJ, Prosser JI, Schimel JP, Whitman WB et al. (2010). The ecological coherence of high bacterial taxonomic ranks. Nat Rev Microbiol 8: 523–529.
Qiu Y-L, Muramatsu M, Hanada S, Kamagata Y, Guo R-B, Sekiguchi Y . (2013). Oligosphaera ethanolica gen. nov., sp. nov., an anaerobic, carbohydrate-fermenting bacterium isolated from methanogenic sludge, and description of Oligosphaeria classis nov. in the phylum Lentisphaerae. Int J Syst Evol Microbiol 63: 533–539.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41: D590–D596.
Reeves RE . (1971). Pyruvate,phosphate dikinase from Bacteroides symbiosus. Biochem J 125: 531–539.
Santarella-Mellwig R, Pruggnaller S, Roos N, Mattaj IW, Devos DP . (2013). Three-dimensional reconstruction of bacteria with a complex endomembrane system. PLoS Biol 11: e1001565.
Schneider D, Arp G, Reimer A, Reitner J, Daniel R . (2013). Phylogenetic analysis of a microbialite-forming microbial mat from a hypersaline lake of the Kiritimati atoll, Central Pacific. PLoS One 8: e66662.
Seemann T . (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069.
Spring S, Brinkmann N, Murrja M, Spröer C, Reitner J, Klenk H-P . (2015). High diversity of culturable prokaryotes in a lithifying hypersaline microbial mat. Geomicrobiol J 32: 332–346.
Spring S, Riedel T, Spröer C, Yan S, Harder J, Fuchs BM . (2013). Taxonomy and evolution of bacteriochlorophyll a-containing members of the OM60/NOR5 clade of marine gammaproteobacteria: description of Luminiphilus syltensis gen. nov., sp. nov., reclassification of Haliea rubra as Pseudohaliea rubra gen. nov., comb. nov. BMC Microbiol 13: 118.
Spring S, Scheuner C, Lapidus A, Lucas S, Glavina Del Rio T, Tice H et al. (2010). The genome sequence of Methanohalophilus mahii SLPT reveals differences in the energy metabolism among members of the Methanosarcinaceae inhabiting freshwater and saline environments. Archaea 2010: Article ID 690737.
Steelman SM, Chowdhary BP, Dowd S, Suchodolski J, Janečka JE . (2012). Pyrosequencing of 16S rRNA genes in fecal samples reveals high diversity of hindgut microflora in horses and potential links to chronic laminitis. BMC Vet Res 8: 231.
Steer T, Collins MD, Gibson GR, Hippe H, Lawson PA . (2001). Clostridium hathewayi sp. nov., from human faeces. Syst Appl Microbiol 24: 353–357.
Tailford LE, Crost EH, Kavanaugh D, Juge N . (2015). Mucin glycan foraging in the human gut microbiome. Front Genet 6: 81.
Thorvaldsdóttir H, Robinson JT, Mesirov JP . (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192.
Tindall BJ . (1990). Lipid composition of Halobacterium lacusprofundi. FEMS Microbiol Lett 66: 199–202.
Tindall BJ, Sikorski J, Smibert RA, Krieg NR. (2007) Phenotypic characterization and the principles of comparative systematic. In: Reddy C, Beveridge T, Breznak J, Marzluf G, Schmidt T, Snyder L (eds), Methods for General and Molecular Microbiology, 3rd edn. American Society of Microbiology: Washington, DC, USA, pp 330–393..
Van Mooy BA, Fredricks HF, Pedler BE, Dyhrman ST, Karl DM, Koblízek M et al. (2009). Phytoplankton in the ocean use non-phosphorus lipids in response to phosphorus scarcity. Nature 458: 69–72.
Waack S, Keller O, Asper R, Brodag T, Damm C, Fricke WF et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7: 142.
Wagner M, Horn M . (2006). The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr Opin Biotechnol 17: 241–249.
Wang Q, Garrity GM, Tiedje JM, Cole JR . (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267.
Wegner CE, Richter-Heitmann T, Klindworth A, Klockow C, Richter M, Achstetter T et al. (2013). Expression of sulfatases in Rhodopirellula baltica and the diversity of sulfatases in the genus. Rhodopirellula Mar Genomics 9: 51–61.
Wittmann J, Dreiseikelmann B, Rohde M, Meier-Kolthoff JP, Bunk B, Rohde C . (2014). First genome sequences of Achromobacter phages reveal new members of the N4 family. Virol J 11: 14.
Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer K-H et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12: 635–645.
Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C et al. (2014). The SILVA and ‘all-species Living Tree Project (LTP)’ taxonomic frameworks. Nucleic Acids Res 42: D643–D648.
Yilmaz P, Yarza P, Rapp JZ, Glöckner FO . (2016). Expanding the world of marine bacterial and archaeal clades. Front Microbiol 6: 1524.
Zoetendal EG, Plugge CM, Akkermans ADL, de Vos WM . (2003). Victivallis vadensis gen. nov., sp. nov., a sugar-fermenting anaerobe from human faeces. Int J Syst Evol Microbiol 53: 211–215.
Acknowledgements
We thank Anja Frühling, Gabriele Pötter and Anika Wasner for help in the chemotaxonomical analyses; Simone Severitt and Nicole Mrotzek for excellent technical assistance regarding SMRTbell template preparation and sequencing; and Sabine Welnitz for preparing biomass of Akkermansia muciniphila DSM 22959T and Spirochaeta dissipatitropha DSM 23605T. We are grateful to the Genome Analytics group (HZI Braunschweig) for providing Illumina sequence data. Three anonymous reviewers are acknowledged for their helpful comments. This study has been funded by the German Research Foundation, project Kl 1000/2-2, and the Leibniz Institute DSMZ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Spring, S., Bunk, B., Spröer, C. et al. Characterization of the first cultured representative of Verrucomicrobia subdivision 5 indicates the proposal of a novel phylum. ISME J 10, 2801–2816 (2016). https://doi.org/10.1038/ismej.2016.84
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2016.84
This article is cited by
-
Longitudinal study of the short- and long-term effects of hospitalisation and oral trimethoprim-sulfadiazine administration on the equine faecal microbiome and resistome
Microbiome (2023)
-
Cefquinome shows a higher impact on the pig gut microbiome and resistome compared to ceftiofur
Veterinary Research (2023)
-
Verrucomicrobiota are specialist consumers of sulfated methyl pentoses during diatom blooms
The ISME Journal (2022)
-
Sulfuriroseicoccus oceanibius gen. nov., sp. nov., a representative of the phylum Verrucomicrobia with a special cytoplasmic membrane
Antonie van Leeuwenhoek (2022)
-
Characterization of oral and cloacal microbial communities of wild and rehabilitated loggerhead sea turtles (Caretta caretta)
Animal Microbiome (2021)
