
Abstract
Mycoplankton have so far been a neglected component of pelagic marine ecosystems, having been poorly studied relative to other plankton groups. Currently, there is a lack of understanding of how mycoplankton diversity changes through time, and the identity of controlling environmental drivers. Using Fungi-specific high-throughput sequencing and quantitative PCR analysis of plankton DNA samples collected over 6 years from the coastal biodiversity time series site Station L4 situated off Plymouth (UK), we have assessed changes in the temporal variability of mycoplankton diversity and abundance in relation to co-occurring environmental variables. Mycoplankton diversity at Station L4 was dominated by Ascomycota, Basidiomycota and Chytridiomycota, with several orders within these phyla frequently abundant and dominant in multiple years. Repeating interannual mycoplankton blooms were linked to potential controlling environmental drivers, including nitrogen availability and temperature. Specific relationships between mycoplankton and other plankton groups were also identified, with seasonal chytrid blooms matching diatom blooms in consecutive years. Mycoplankton α-diversity was greatest during periods of reduced salinity at Station L4, indicative of riverine input to the ecosystem. Mycoplankton abundance also increased during periods of reduced salinity, and when potential substrate availability was increased, including particulate organic matter. This study has identified possible controlling environmental drivers of mycoplankton diversity and abundance in a coastal sea ecosystem, and therefore sheds new light on the biology and ecology of an enigmatic marine plankton group. Mycoplankton have several potential functional roles, including saprotrophs and parasites, that should now be considered within the consensus view of pelagic ecosystem functioning and services.
Similar content being viewed by others


Introduction
Plankton are the foundation of marine ecosystems because they sustain food webs and drive global biogeochemical cycles (Worden et al., 2015). Some plankton groups, such as phytoplankton and zooplankton, have been studied extensively for decades, with earlier research founded on microscope-based assessments of diversity and abundance (Russell et al., 1971; Southward, 1974). More recently, through the application of DNA-based tools that incorporate previously poorly described plankton groups, such as bacterioplankton and protists, marine plankton diversity has been shown to be vast, complex and influenced by a wide range of environmental drivers (Caporaso et al., 2012; Logares et al., 2014; de Vargas et al., 2015).
Even though planktonic fungi (mycoplankton) have been known to be present in marine ecosystems for several decades (Vishniac, 1956), they have received considerably less attention compared with other plankton groups (Wang et al., 2012). Marine mycoplankton exist as free-living filamentous and yeast forms, or as parasites of other plankton (Richards et al., 2012; Wang et al., 2012). The ecosystem functioning roles that marine mycoplankton may undertake have been largely assumed from comparison with their terrestrial and freshwater analogues (Richards et al., 2012). For example, Richards et al. (2012) proposed that, similar to terrestrial fungi, saprotrophic marine fungi probably attach to particulate organic material, produce extracellular enzymes and utilise resulting lower molecular weight compounds. Supporting this theory, evidence from the coastal waters off Chile suggest that mycoplankton have a major role in processing phytoplankton-derived biopolymers using extracellular enzymes (Gutiérrez et al., 2011).
Relative to other plankton groups, such as the bacterioplankton, there have been a limited number of assessments of marine mycoplankton diversity. So far, studies of marine mycoplankton diversity have been largely based on samples collected from one-off sampling campaigns that have provided important and insightful ‘snap shots’ of diversity, but with limited temporal resolution (Bass et al., 2007; Gao et al., 2010; Richards et al., 2012; Manohar and Raghukumar, 2013; Taylor and Cunliffe, 2014). Time series assessments of other plankton groups have proven invaluable in developing our understanding of their biology and ecology (Karl and Church, 2014; Fuhrman et al., 2015). Spatially defined and reoccurring assessments of plankton diversity and abundance that are aligned with co-occurring environmental variables (temperature, nutrients and so on) have been used to identify the key controlling environmental drivers that influence planktonic organisms, and to form the basis of establishing the underpinning principles of their ecologies and ecosystem functional roles. For example, multi-year assessments of bacterioplankton diversity at the time series site Station L4 in the Western English Channel has shown that bacterioplankton have annually repeating seasonal-scale patterns in diversity that relate to changes in a variety of environmental drivers, including seawater temperature, seasonal diatom blooms and organic carbon availability (Gilbert et al., 2009, 2012; Taylor et al., 2014).
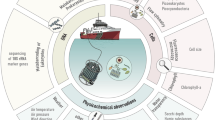
The coastal time series site Station L4 is located 13 km off Plymouth in the Western English Channel and has a depth of 50 m (Figure 1). The site is dynamic and influenced by periodic estuarine incursions from the neighbouring Tamar and Plym rivers. During the summer, the water column becomes thermally stratified at Station L4 and nutrient depletion occurs (Smyth et al., 2010). Here we present a multi-year time series assessment of mycoplankton diversity and abundance, correlating observed changes with co-occurring environmental parameters. We have focussed on the mycoplankton communities at Station L4 because previous studies have indicated that mycoplankton diversity is greater in coastal waters than in the open ocean (Gao et al., 2010; Wang et al., 2014), and the station has been proven to be an exemplar site for elucidating the ecology and biology of other plankton groups (Eloire et al., 2010; Widdicombe et al., 2010; Gilbert et al., 2012).

Location of Station L4 in the Western English Channel.
Materials and methods
Seawater sampling and environmental metadata
Seawater samples were collected as part of the regular sampling at the Western Channel Observatory (WCO) site Station L4 (50° 15.0' N 4° 13.2' W; Figure 1; www.westernchannelobservatory.org.uk) between 2008 and 2013 (Supplementary Table 1). Three litres of surface sea water (3 m depth) was filtered through 0.2 μm Sterivex filter units (Millipore, Darmstadt, Germany) and the filters stored at −80 °C.
The environmental metadata used in this study were collected at the same time as the seawater samples for DNA extraction (above) also as part of the regular activities at the WCO (Table 1 and Supplementary Figure 1). Temperature and salinity were determined via a rosette-mounted SeaBird SBE 19+ CTD (Sea-Bird Electronics, St Bellevue, WA, USA). Nutrients were measured in seawater samples collected via a sampling bottle attached to the rosette and using established protocols for nitrate (Brewer and Riley, 1965), nitrite (Grasshoff et al., 1999), ammonia (Mantoura and Woodward, 1983), phosphate (Zhang and Chi, 2002) and silicate (Kirkwood, 1989). The concentrations of particulate organic carbon, total particulate nitrogen and particulate organic nitrogen were determined by filtering 250 ml seawater samples (prefiltered through 200 μm mesh) onto ashed glass fibre filters (GF/F) and analysed using an FlashEA 1112 elemental analyser (Thermo Scientific, Loughborough, UK). For chlorophyll a determination, 100 ml of sea water was filtered onto GF/F, followed by extraction in 90% acetone at 4 °C, and fluorescence measured using a fluorometer (Welschmeyer, 1994). To determine diatom abundance and diversity, 200 ml seawater samples were fixed with Lugol’s iodine solution (2% final concentration) and examined by light microscopy via the Utermöhl counting technique (Widdicombe et al., 2010). Metadata are available from the WCO (www.westernchannelobservatory.org.uk).
DNA extraction, PCR and sequencing
Filters were transferred to 2 ml microcentrifuge tubes and 1 ml phenol/chloroform/isoamyl alcohol (25:24:1) was added. The tubes were vortexed for 2 min, heated for 10 min at 60 °C, before being vortexing again and centrifuged for 1 min at 13 000 g. The aqueous phase (~0.5 ml) was transferred to a new 1.5 ml tube containing 0.5 ml chloroform/isoamyl alcohol, briefly vortexed and centrifuged again for 1 min at 13 000 g. The aqueous phase was transferred again to a new 1.5 ml microcentrifuge tube followed by 10% (v/v) 3 M sodium acetate and 2 volumes of molecular-grade ethanol. DNA was precipitated for 1 h at −20 °C, followed by centrifugation at 13 000 g for 25 min. The supernatant was removed and the DNA pellet washed with 0.5 ml 75% ethanol, before being dissolved in 50 μl nuclease free molecular-grade water and stored at −80 °C.
The fungi-specific internal transcribed spacer (ITS) region (ITS1,5.8S, ITS2) was amplified using the PCR primers ITSF 5′-CTTGGTCATTTAGAGGAAGTAA-3′ and ITS4 5′-TCCTCCGCTTATTGATATGC-3′ (White et al., 1990), including a barcode on the forward primer and the HotStarTaq Plus Master Mix Kit (Qiagen, Manchester, UK) according to the manufacturer's instructions. The following PCR regime was used: 94 °C for 3 min, followed by 28 cycles of 94 °C for 30 s, 53 °C for 40 s and 72 °C for 1 min, and a final elongation step at 72 °C for 5 min. PCR products were assessed by agarose gel electrophoresis. Three separate PCR amplifications were performed from each DNA sample, and were subsequently pooled for sequencing. All sample processing was undertaken with molecular-grade reagents and in a clean laminar flow PCR hood, including exposure of fungal growth plates to assess potential aerial contamination. We also incorporated negative controls throughout all steps in our analysis and found no fungal contamination.
The pooled samples were purified using calibrated Ampure XP beads (Beckman Coulter, High Wycombe, UK) and used to prepare DNA libraries by following the Illumina TruSeq DNA library preparation protocol (Illumina, Inc., California, CA, USA). Sequencing was performed using the Illumina MiSeq platform following the manufacturer’s guidelines.
Bioinformatics
Sequences were analysed using a combination of USEARCH v7.0.1090 (32Bit) (Edgar, 2010) and QIIME v1.8.0 (Caporaso et al., 2010). Barcodes were removed from multiplexed fastq files using USEARCH python command script fastq_strip_barcode_relabel2.py. Fastq files were quality filtered (low quality=expected error >0.5 and short sequences <250 bp), sequence length truncated to 250 bp and converted to FASTA files. The FASTA files were de-replicated, abundance sorted and singleton sequences were removed. Operational taxonomic units (OTUs) were clustered de novo using the UPARSE clustering algorithm (Edgar, 2013). Chimeras were then filtered using UCHIME (Edgar et al., 2011) and the UNITE 97% OTU library (version 12_11) (Abarenkov et al., 2010) as a reference. OTUs were then mapped back to the original reads and an OTU table produced. Because marine fungi are not relatively well characterised and databases for fungal ITS sequences are by no means complete, extra care was taken in the annotation of sequences. First, taxonomy was assigned to OTUs using the BLAST method in QIIME and against three specialist fungal databases: the UNITE database (Abarenkov et al., 2010), a fungal metagenomics ITS database (Taylor and Houston, 2011) and the ITSone database (Santamaria et al., 2012). Results were cross compared and correct annotation chosen on agreement between the databases. When agreement was not reached, the highest query coverage and percentage ID was chosen as a match. To accurately identify metazoa, other eukaryote sequences and confirm taxonomy, the data set was BLAST searched against the full NCBI (National Center for Biotechnology Information) nucleotide reference database. After taxonomy assignment, singletons were removed using the script filter_otus_from_otu_table.py and metazoans were removed from the data set using the script filter_taxa_from_otu_table.py. The script alpha_diversity.py was used to calculate species richness and diversity indices. Sequences are available from the European Nucleotide Archive (accession code PRJEB9844).
Fungi-specific 18S rRNA gene quantitative PCR (Q-PCR)
Fungi-specific Q-PCR primers FR1 5′-AICCATTCAATCGGTAIT-3′ and FF390 5′-CGATAACGAACGAGACCT-3′ (Prevost-Boure et al., 2011) were used with the Sensi-FAST SYBR Q-PCR kit (Bioline, London, UK). The 10 μl reactions contained 5 μl Sensi-FAST master mix, 0.25 μl of each primer (final concentration 0.4 μM), 1 μl of DNA template (~50 ng DNA) and 4 μl nuclease-free molecular-grade water. A Qiagen Rotor-Gene (Qiagen) was used to perform Q-PCR. The following Q-PCR regime was used: 94 °C for 3 min, then 40 cycles of 94 °C for 10 s, annealing at 50 °C for 15 s and elongation and acquisition of fluorescence data at 72 °C for 20 s. Standard curves were constructed using known amounts of target template generated by PCR amplification of the target gene from genomic DNA from Saccharomyces cerevisiae. The resulting amplicon was purified using a Gen-Elute PCR purification kit (Sigma, Dorset, UK) before quantification on a Nanodrop ND-1000 spectrophotometer (Nanodrop, Loughborough, UK).
Statistical analysis
Correlations between changes in OTU abundance and co-occurring environmental metadata were determined using Pearson’s correlation analysis in QIIME using the script observation_metadata_correlation.py. Correlations between changes in α-diversity parameters (numbers of OTUs and Shannon Index) and Fungi 18S rRNA gene abundance with co-occurring environmental metadata were determined using Spearman’s rank correlation analysis in the statistical software package SPSS (IBM Corporation, Portsmouth, UK).
Results
Abundant and dominant mycoplankton at Station L4
Fungi ITS gene libraries generated from plankton DNA samples collected from surface waters at Station L4 were principally composed of the phyla Ascomycota, Basidiomycota and Chytridiomycota. Within these three phyla, several orders were prevalent across the 6-year time series (Figure 2a) that were frequently abundant (1–50% of sequences in libraries) and sometimes dominant (50–100% of sequences in libraries) within individual plankton samples (Figure 2b). We subsequently focussed on the distribution of these potential bloom-forming mycoplankton orders that reoccurred in multiple years across the time series at Station L4 (Figure 3). The nine bloom-forming orders accounted for an average of 70% of the total sequences in the Fungi ITS gene libraries generated, and were each composed of multiple OTUs, except for the unclassified Chytridiomycota that was made up of a single OTU (Figure 4).

(a) Averaged normalised abundance of mycoplankton orders in plankton samples collected from surface waters at Station L4 in the Western English Channel from January 2008 to June 2013. Orders <0.1% have been omitted from the figure. (b) Frequency of mycoplankton orders in individual plankton samples classified as abundant (1–49% of sequences in libraries; light colour) and dominant (50–100% of sequence libraries; dark colour). The arrows highlight mycoplankton orders that frequently formed blooms and that were studied in more detail.

Change in the normalised abundance of bloom-forming mycoplankton orders in surface waters at Station L4 in the Western English Channel identified in Figure 2. The data shown are normalised against total reads at each time point.

Distribution of abundant (1–49% of sequences in libraries; light colour) and dominant (50–100% of sequence libraries; dark colour) OTUs within the bloom-forming mycoplankton orders in Figure 3.
Changes in the abundance of interannually reoccurring OTUs (that is, OTUs abundant or dominant in at least two different years) were compared with changes in cognate environmental parameters (Table 2 and Supplementary Figure 1). Reoccurring OTUs in the Ascomycota orders Capnodiales and Dothideales, and the Basidiomycota orders Geastrales and Malasseziales, were increased in abundance at Station L4 when the concentrations of nitrogen-containing compounds were also increased (Table 2). Changes in salinity were linked to several Capnodiales OTUs that were increased in abundance during periods of reduced salinity. Interesting, the salinity-responding OTUs showed closest sequence similarities to other environmentally derived sequences and fungal isolates associated with land plants (Table 3). Seasonal increases in seawater temperature were linked to increases in abundance of some Ascomycota and Basidiomycota OTUs. Conversely, OTU1 (order Dothideales) was most abundant when the sea water was cooler at Station L4, and also when phosphate concentrations were increased. The Chytridiomycota OTU14 formed blooms in June 2008, July 2009 and June 2011 that coincided with seasonal increases in diatom abundance (Table 2 and Figure 5). The diatom communities during the OTU14 blooms were dominated by Chaetoceros spp. and Attheya septentrionalis in 2008, Chaetoceros spp. in 2009 and Chaetoceros spp. and Proboscia alata in 2011 (Supplementary Figure 2).

Change in the normalised relative abundance of the Chytrid OTU14 and diatom abundance determined from Utermöhl chamber light-microscope cell counts in plankton samples collected from surface waters at Station L4 in the Western English Channel from January 2008 to June 2013. The data shown are normalised against total reads at each time point.
Mycoplankton α-diversity
Mycoplankton α-diversity was measured by determining the number and distribution of OTUs within individual Fungi ITS gene libraries generated from the plankton DNA samples. The number of OTUs and OTU richness, determined as the Shannon Index, varied considerably between the samples, and between the three major phyla in the study (Table 4). Changes in the total Fungi Shannon Index correlated with changes in salinity (n=35; P=0.03; rho=−0.367), with diversity greatest during periods when salinity was reduced (Figure 6a).

(a) Relationship between the total Fungi Shannon Index of individual ITS gene libraries (2000 sequences per library) and salinity at Station L4. (b) Relationship between the number of Ascomycota OTUs and (c) Ascomycota Shannon Index within individual ITS gene libraries (500 sequences per library) and salinity at Station L4. (d) Relationship between Fungi 18S rRNA gene copies and salinity at Station L4. The dashed lines represent the linear regression.
Focussing on the three major phyla showed that there was also a significant link between the number of Ascomycota OTUs, Ascomycota Shannon Index and salinity (OTUs n=30; P<0.01; rho=−0.617; Shannon n=30; P<0.01; rho=−0.628), with Ascomycota diversity greatest at lower salinities (Figures 6b and c). Changes in Ascomycota diversity also coincided with changes in the concentration of ammonia (OTUs n=26; P=0.03; rho=0.426; Shannon n=26; P=0.02; rho=0.456) and phosphate (OTUs n=24; P=0.04; rho=0.423), with diversity greatest when ammonia and phosphate concentrations were increased (Supplementary Table 2). Changes in Basidiomycota α-diversity across the time series could not be linked to changes in the co-occurring environmental parameters at Station L4. Chytridiomycota diversity and number of associated samples were too low for comparable analysis.
Mycoplankton abundance
Mycoplankton abundance was estimated by using Fungi-specific 18S rRNA gene Q-PCR analysis of plankton DNA samples, and showed that Fungi 18S rRNA genes varied from 5.1 × 105 to 9.9 × 107 copies l−1 over the study period at Station L4 (Supplementary Figure 3). It important to reiterate that 18S rRNA genes were used as an estimate of mycoplankton abundance and that different taxa may have varying copies of the gene. Mycoplankton abundance was linked to fluctuations in salinity (n=46; P=0.03; rho=−0.327), with abundance highest during periods when salinity was reduced (Figure 6d).
Changes in mycoplankton abundance also showed significant links to changes in the concentrations of particulate organic carbon (n=42; P=0.03; rho=0.323), ammonia (n=43; P<0.01; rho=0.471), total particulate nitrogen (n=42; P<0.01; rho=0.510) and particulate organic nitrogen (n=28; P<0.01; rho=0.489) (Supplementary Table 2). In all cases, increases in mycoplankton abundance coincided with the increase in the concentration of the environmental parameters.
Discussion
In this multi-year time series study we show that coastal mycoplankton diversity is dominated by the Dikarya phyla Ascomycota and Basidiomycota, and to a lesser extent the phylum Chytridiomycota. This view of fungal diversity is consistent with other culture-independent studies of different marine ecosystems (Richards et al., 2012; Manohar and Raghukumar, 2013), including the surface open ocean (Wang et al., 2014), the deep open ocean (Bass et al., 2007), deep ocean sediments (Orsi et al., 2013a) and the microbiomes of other marine organisms (Wegley et al., 2007).
The changes in mycoplankton abundance throughout the time series at Station L4, estimated from Fungi-specific 18S rRNA gene Q-PCR analysis, indicate that the mycoplankton assemblages in the water column of the English Channel are dynamic and are not stable. Other studies have quantified temporal changes in the abundance of different plankton groups at Station L4 and have shown similar dynamic fluctuations. Bacterioplankton, which are dominated at Station L4 by Bacteria (Archaea <15%), have been shown in one annual cycle to range from 0.2 × 106 to 1.6 × 106 cells ml−1 using flow cytometry and tyramide signal amplification–fluorescent in situ hybridisation (Mary et al., 2006). Quantification of specific groups of phytoplankton (dinoflagellates, coccolithophores, diatoms and so on) using light microscopy have also shown that substantial changes in abundance occur (Widdicombe et al., 2010). At present, it is not possible to make direct carbon biomass estimates from the Fungi 18S rRNA gene Q-PCR-derived abundance data produced in this study; however, because abundance changed rapidly, it is likely that rapid carbon turnover is taking place. Gutiérrez et al. (2011) have shown that in the upwelling region off Chile, mycoplankton biomass can reach similar levels to bacterioplankton biomass. This suggests that mycoplankton biomass is potentially a significant and dynamic component of the coastal carbon cycle.
Several studies have shown that fungi are abundant in marine sediments (Orsi et al., 2013a, 2013b; Xu et al., 2014; Taylor and Cunliffe, 2015). Xu et al. (2014) used a similar Q-PCR-based approach to quantify fungi in sediment samples collected from five deep-sea sites in the Pacific Ocean and showed that Fungi-specific 28S rRNA gene copies ranged from 3.5 × 106 to 5.2 × 107 copies per g wet sediment (Xu et al., 2014). Assuming that 1 g sediment≈1 ml seawater, then as with bacteria in sediments and bacterioplankton, fungi in sediments occur at higher abundances than mycoplankton in the water column.
At Station L4, episodic reductions in salinity were linked to the increased abundance of bloom-forming Capnodiales OTUs (Ascomycota), increased total Fungi and Ascomycota α-diversity, and increased mycoplankton abundance determined by Q-PCR. Station L4 is located 13 km from the coast, and is periodically affected by riverine inputs from the Tamar and Plym estuaries caused by variations in the tide, rainfall and wind mixing, and which result in the short-term freshening of surface sea water (Smyth et al., 2010). It is possible that the significant changes in mycoplankton diversity and abundance in relation to the reductions in salinity at Station L4 are caused by fungi being transported from the neighbouring riverine systems to the coastal station. Because fungi have robust chitin-rich cell walls, it is likely that mycoplankton can withstand the osmotic shock caused by the transition from fresh and brackish waters into seawater (Richards et al., 2012).
Burgaud et al. (2013) performed a transect study of benthic and planktonic fungi at stations along the Delaware River estuary using clone library analysis, cultivation and fluorescent in situ hybridisation, and showed that diversity and abundance decreased along the estuarine transect from the freshwater stations to the marine station (salinity range 0.08–31.5), concluding that salinity has an important impact on fungi. A similar clone library-based study of sediment fungi at three sites along a saltmarsh salinity gradient (salinity range 0–33) also concluded that salinity, along with co-occurring variables such as changes in plant species, has an significant impact on fungal diversity (Mohamed and Martiny, 2011). As we propose here for Station L4, the Delaware River study also suggested that some fungi that are present in coastal marine ecosystems are terrestrially derived and are transported there by rivers (Burgaud et al., 2013).
Riverine inputs of fungi to coastal waters may to some extent account for the observations made of the biogeographical distribution of mycoplankton in the Pacific Ocean that have shown that diversity is greater in coastal waters around the Hawaiian Islands compared with the open ocean (Gao et al., 2010; Wang et al., 2014), and that Ascomycota in particular are especially more abundant nearer the coast (Wang et al., 2014).
Coastal ecosystems with regular riverine inputs of mycoplankton may also have a role in the longer-term radiation of marine fungal diversity by facilitating terrestrial–marine transitions. Through multi-phyla phylogenetic analysis, Richards et al. (2012) have proposed that multiple marine–terrestrial fungal transitions have occurred, and that novel marine lineages have developed. Specific phylogenetic analyses focussed on cultivated representatives of marine Ascomycota lineages, including the order Capnodiales, also suggest that multiple marine–terrestrial transitions have taken place (Spatafora et al., 1998; Suetrong et al., 2009). Cultured marine representatives of the Ascomycota class Dothideomycetes, which includes some of the bloom-forming orders reported in this study (for example, Pleosporales and Dothideales), have morphological adaptions to the marine environment that support effective discharge of ascospores, such as protective mucilaginous sheaths (Suetrong et al., 2009). The Delaware River study found that most fungi isolated from the ecosystem were halotolerant (Burgaud et al., 2013). It is possible that terrestrial and freshwater fungi transported from the river catchment that survive in coastal waters may subsequently be dispersed to open ocean waters.
Mycoplankton diversity and abundance at Station L4 was also affected by changes in the availability of several of potential growth substrates. Coastal ecosystems are typically highly productive because of elevated levels of autochthonous production and allochthonous inputs. Mycoplankton abundance determined by microscopy in the productive coastal upwelling waters off Chile has been linked to increased availability of autochthonous phytoplankton-derived particulate material (Gutiérrez et al., 2011). Similarly, we show that at Station L4 both mycoplankton diversity and abundance are linked to changes in availability of particulate organic matter. Active fungal communities within deep sub-seafloor sediments also show similar positive links to changes in the availability of organic material (Orsi et al., 2013a), and the expression of metabolic pathways involved in the cycling of a variety of organic substrates (Orsi et al., 2013b). The utilisation of particulate organic matter by benthic and pelagic marine fungi is analogous to the well-established functional roles of fungi in freshwater ecosystems, such as lakes (Wurzbacher et al., 2010).
Changes in organic and inorganic nitrogen-rich substrates had particular impact on abundance and diversity at Station L4. Metagenomic analysis of microbial communities associated with the coral Porites astreoides has shown a high prevalence of fungal genes involved in nitrogen metabolism, including ammonia assimilation and nitrite ammonification (Wegley et al., 2007). Nitrite reduction by fungi in deep sub-seafloor sediments has also been proposed (Orsi et al., 2013b). Changes in substrate availability and associated niche differentiation could be one mechanism through which saprotrophic mycoplankton diversity and abundance is controlled in pelagic ecosystems.
Chytrids are zoosporic fungi that are either saprotrophs or parasites. The group are relatively well characterised in terrestrial and freshwater ecosystems; however, marine chytrids are poorly understood (Gleason et al., 2011), including the interactions between marine chytrids and diatoms (Scholz et al., 2016). Because of the significant coupling between OTU14 abundance and seasonal diatom blooms at Station L4, it is possible that the chytrid is a diatom parasite. Infections of marine diatoms by nonfungal parasites have already been shown in plankton communities in situ. For example, large-scale infections of the diatoms Guinardia delicatula and G. flaccida by the parasites Pirsonia and Cryothecomonas, both in the phylum Cercozoa, have been reported in the Wadden Sea, with up to 35% of G. delicatula and 65% of G. flaccida infected (Tillmann et al., 1999). A multi-year study of G. delicatula populations on the New England Shelf showed that infection by Cryothecomonas-like parasites regularly exceeded 10% of the population, and that parasite infection appeared to have a negative impact on diatom bloom development (Peacock et al., 2014).
We did not observe increased abundance of OTU14 in 2012 at Station L4, even though diatom blooms still occurred. The 2012 diatoms blooms at the same time in the year when previously OTU14 was abundant were dominated by different diatom species (that is, Leptocylindrus spp. and Guinardia striata) compared with the other years in the study (Supplementary Figure 2). Chytrids are presumed to be host specific, but the relationship is not yet fully understood (Kagami et al., 2007). Dinomyces arenysensis is a chytrid parasite of dinoflagellates that was originally isolated from an Alexandrium minutum bloom, and subsequently shown in laboratory assays with different phytoplankton species to have a broad host range but generally within the genus Alexandrium (Lepelletier et al., 2014). Change in diatom diversity at Station L4 may account for the loss of the chytrids in 2012. It is important to note that no other abundant Chytridiomycota OTUs were recorded in 2012 at Station L4.
Freshwater diatoms have been shown to defend against chytrid infections via a range of mechanisms (Kagami et al., 2007). Analogous explanations for the loss of the Chytrid OTU14 in 2012 at Station L4 include diatom hosts developing anti-parasite defense mechanisms, such as the production of harmful chemicals (Pohnert et al., 2007), or changes within host populations towards more parasite-resistant genotypes (Gsell et al., 2013). Further studies are clearly needed to explore the relationships between diatoms and chytrids at Station L4, and the potential impacts of diatom–chytrid interaction on coastal ecosystem functioning.
Conclusions
Through a multi-year analysis of planktonic marine fungi we have shown that mycoplankton abundance and diversity are variable in coastal waters, and that some of this variation can be accounted for by fluctuating co-occurring ecological conditions, such as salinity, seawater temperature, substrate availability and potential host abundance. Several mycoplankton taxa from different phyla form interannually reoccurring blooms that can to a large extent be explained by specific environmental drivers. We propose that riverine inputs have an important impact on coastal mycoplankton abundance and diversity, with potential implications for the diversity of marine mycoplankton more generally. Our data empirically reinforce the developing paradigm that, similar to terrestrial and freshwater fungi, marine mycoplankton occupy a complex range of niches as saprotrophs, nutrient recyclers and parasites. Mycoplankton must be considered within an overall view of coastal pelagic ecosystem structure and function.
Accession codes
References
Abarenkov K, Henrik Nilsson R, Larsson K, Alexander I, Eberhardt U, Erland S et al. (2010). The UNITE database for molecular identification of fungi—recent updates and future perspectives. New Phytol 189: 281–285.
Bass D, Howe A, Brown N, Barton H, Demidova M, Michelle H et al. (2007). Yeast forms dominate fungal diversity in the deep oceans. Proc R Soc B 274: 3069–3077.
Brewer PG, Riley JP . (1965). The automatic determination of nitrate in sea water. Deep-Sea Res 12: 765–772.
Burgaud G, Woehlke S, Rédou V, Orsi W, Beaudoin D, Barbier G et al. (2013). Deciphering the presence and activity of fungal communities in marine sediments using a model estuarine system. Aquat Microb Ecol 70: 45–62.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Caporaso JG, Paszkiewicz K, Field D, Knight R, Gilbert JA . (2012). The Western English Channel contains a persistent microbial seed bank. ISME J 6: 1089–1093.
de Vargas C, Audic S, Henry N, Decelle J, Mahé F, Logares R et al. (2015). Eukaryotic plankton diversity in the sunlit ocean. Science 348: 1261605.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R . (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194–2200.
Edgar RC . (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10: 996–998.
Eloire D, Somerfield PJ, Conway DVP, Halsband-Lenk C, Harris R, Bonnet D . (2010). Temporal variability and community composition of zooplankton at station L4 in the Western Channel: 20 years of sampling. J Plankton Res 32: 657–679.
Fuhrman JA, Cram JA, Needham DM . (2015). Marine microbial community dynamics and their ecological interpretation. Nat Rev Microbiol 13: 133–146.
Gao Z, Johnson ZI, Wang G . (2010). Molecular characterization of the spatial diversity and novel lineages of mycoplankton in Hawaiian coastal waters. ISME J 4: 111–120.
Gilbert JA, Field D, Swift P, Newbold L, Oliver A, Smyth T et al. (2009). The seasonal structure of microbial communities in the Western English Channel. Environ Microbiol 11: 3132–3139.
Gilbert JA, Steele JA, Caporaso JG, Steinbrück L, Reeder J, Temperton B et al. (2012). Defining seasonal marine microbial community dynamics. ISME J 6: 298–308.
Gleason FH, Küpper FC, Amon JP, Picard K, Gachon CMM, Marano AV et al. (2011). Zoosporic true fungi in marine ecosystems: a review. Mar Freshw Res 62: 383–393.
Grasshoff K, Kremling K, Ehrhardt M, Anderson LG . (1999) Methods of Seawater Analysis. Wiley-VCH: Weinheim.
Gsell AS, de Senerpont Domis LN, Verhoeven KJ, van Donk E, Ibelings BW . (2013). Chytrid epidemics may increase genetic diversity of a diatom spring-bloom. ISME J 7: 2057–2059.
Gutiérrez MH, Pantoja S, Tejos E, Quiñones RA . (2011). The role of fungi in processing marine organic matter in the upwelling ecosystem off Chile. Mar Biol 158: 205–219.
Kagami M, Bruin Ad, Ibelings BW, Donk EV . (2007). Parasitic chytrids: their effects on phytoplankton communities and food-web dynamics. Hydrobiologia 578: 113–129.
Karl DM, Church MJ . (2014). Microbial oceanography and the Hawaii Ocean Time-series programme. Nat Rev Microbiol 12: 699–713.
Kirkwood DS . (1989). Simultaneous determination of selected nutrients in seawater. ICES CM C:29, 12.
Lepelletier F, Karpov SA, Alacid E, Le Panse S, Bigeard E, Garcés E et al. (2014). Dinomyces arenysensis gen. et sp. nov. (Rhizophydiales, Dinomycetaceae fam. nov.), a chytrid infecting marine dinoflagellates. Protist 165: 230–244.
Logares R, Audic S, Bass D, Bittner L, Boutte C, Christen R et al. (2014). Patterns of rare and abundant marine microbial eukaryotes. Curr Biol 24: 813–821.
Manohar CS, Raghukumar C . (2013). Fungal diversity from various marine habitats deduced through culture-independent studies. FEMS Microbiol Lett 341: 69–78.
Mantoura RFC, Woodward EMS . (1983). Optimization of the indophenol blue method for the automated determination of ammonia in estuarine waters. Estuar Coast Sheld Sea Sci 17: 219–224.
Mary I, Cummings DG, Biegala IC, Burkill PH, Archer SD, Zubkov MV . (2006). Seasonal dynamics of bacterioplankton community structure at a coastal station in the western English Channel. Aquat Microb Ecol 42: 119–126.
Mohamed DJ, Martiny JB . (2011). Patterns of fungal diversity and composition along a salinity gradient. ISME J 5: 379–388.
Orsi W, Biddle JF, Edgcomb V . (2013a). Deep sequencing of subseafloor eukaryotic rRNA reveals active Fungi across marine subsurface provinces. PLoS One 8: e56335.
Orsi WD, Edgcomb VP, Christman GD, Biddle JF . (2013b). Gene expression in the deep biosphere. Nature 499: 205–210.
Peacock EE, Olson RJ, Sosik HM . (2014). Parasitic infection of the diatom Guinardia delicatula, a recurrent and ecologically important phenomenon on the New England Shelf. Mar Ecol Prog Ser 503: 1–10.
Pohnert G, Steinke M, Tollrian R . (2007). Chemical cues, defence metabolites and the shaping of pelagic interspecific interactions. Trends Ecol Evol 22: 198–204.
Prevost-Boure NC, Christen R, Dequiedt S, Mougel C, Lelievre M, Jolivet C et al. (2011). Validation and application of a PCR primer set to quantify fungal communities in the soil environment by real-time quantitative PCR. PLoS One 6: e24166.
Richards TA, Jones MDM, Leonard G, Bass D . (2012). Marine fungi: their ecology and molecular diversity. Annu Rev Marine Sci 4: 495–522.
Russell FS, Southward AJ, Boalch GT, Butler EI . (1971). Changes in biological conditions in the English Channel off Plymouth during the last half century. Nature 234: 468–470.
Santamaria M, Fosso B, Consiglio A, De Caro G, Grillo G, Licciulli F et al. (2012). Reference databases for taxonomic assignment in metagenomics. Brief Bioinform 13: 682–695.
Scholz B, Guillou L, Marano AV, Neuhauser S, Sullivan BK, Karsten U et al. (2016). Zoosporic parasites infecting marine diatoms – a black box that needs to be opened. Fungal Ecol 19: 59–76.
Smyth TJ, Fishwick JR, AL-Moosawi L, Cummings DG, Harris C, Kitidis V et al. (2010). A broad spatio-temporal view of the Western English Channel observatory. J Plankt Res 32: 585–601.
Southward AJ . (1974). Changes in the plankton community of the Western English Channel. Nature 249: 180–181.
Spatafora JW, Volkmann-Kohlmeyer B, Kohlmeyer J . (1998). Independent terrestrial origins of the Halosphaeriales (marine Ascomycota). Am J Bot 85: 1569–1580.
Suetrong S, Schoch CL, Spatafora JW, Kohlmeyer J, Volkmann-Kohlmeyer B, Sakayaroj J et al. (2009). Molecular systematics of the marine Dothideomycetes. Stud Mycol 64: 155–173.
Taylor DL, Houston S . (2011). A bioinformatics pipeline for sequence-based analyses of fungal biodiversity. In: Xu J-R, Bluhm BH (eds), Fungal Genomics. Springer: Berlin, Germany, pp 141–155.
Taylor JD, Cottingham SD, Billinge J, Cunliffe M . (2014). Seasonal microbial community dynamics correlate with phytoplankton-derived polysaccharides in surface coastal waters. ISME J 8: 245–248.
Taylor JD, Cunliffe M . (2014). High-throughput sequencing reveals neustonic and planktonic microbial eukaryote diversity in coastal waters. J Phycol 50: 960–965.
Taylor JD, Cunliffe M . (2015). Polychaete burrows harbour distinct microbial communities in oil-contaminated coastal sediments. Environ Microbiol Rep 7: 606–613.
Tillmann U, Hesse K-J, Tillmann A . (1999). Large-scale parasitic infection of diatoms in the Northfrisian Wadden Sea. J Sea Res 42: 251–261.
Vishniac HS . (1956). On the ecology of the lower marine fungi. Biol Bull 111: 410–414.
Wang G, Wang X, Liu X, Li Q . (2012) Diversity and biogeochemical function of planktonic fungi in the ocean. In: Raghukumar C (ed), Biology of Marine Fungi. Spinger: Berlin, Germany, pp 71–88.
Wang X, Singh P, Gao Z, Zhang X, Johnson ZI, Wang G . (2014). Distribution and diversity of planktonic fungi in the West Pacific Warm Pool. PLOS One 9: e101523.
Wegley L, Edwards R, Rodriguez-Brito B, Liu H, Rohwer F . (2007). Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ Microbiol 9: 2707–2719.
Welschmeyer NA . (1994). Fluorometric analysis of chlorophyll a in the presence of chlorophyll b and pheopigments. Limnol Oceanogr 39: 1985–1992.
White TJ, Bruns T, Lee S, Taylor J . (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds), PCR Protocols: A Guide to Methods and Applications. Academic Press: San Diego, USA.
Widdicombe CE, Eloire D, Harbour D, Harris RP, Somerfield PJ . (2010). Long-term phytoplankton community dynamics in the Western English Channel. J Plankt Res 32: 643–655.
Worden AZ, Follows MJ, Giovannoni SJ, Wilken S, Zimmerman AE, Keeling PJ . (2015). Rethinking the marine carbon cycle: factoring in the multifarious lifestyles of microbes. Science 347: 1257594.
Wurzbacher CM, Bärlocher F, Grossart HP . (2010). Fungi in lake ecosystems. Aquat Microb Ecol 59: 125–149.
Xu W, Pang KL, Luo ZH . (2014). High fungal diversity and abundance recovered in the deep-sea sediments of the Pacific Ocean. Microb Ecol 68: 688–698.
Zhang JZ, Chi J . (2002). Automated analysis of nanomolar concentrations of phosphate in natural waters with liquid waveguides. Environ Sci Technol 36: 1048–1053.
Acknowledgements
This study was funded by a Marine Biological Association (MBA) Research Fellowship awarded to MC. We thank Declan Schroeder and Cecilia Balestreri (MBA) for kindly providing the plankton genomic DNA samples used in this study from the EU FP7-funded project Marine Microbial Biodiversity, Bioinformatics, Biotechnology (MicroB3). We are indebted to the crews of RV Plymouth Quest and RV Sepia for continued sampling at Station L4. Supporting environmental metadata were provided by the Western Channel Observatory that is funded as part of the UK Natural Environment Research Council’s National Capability. Special thanks to Claire Widdicombe from the Plymouth Marine Laboratory for providing the diatom abundance and diversity data. We also thank three anonymous reviewers for helping to improve on a previous version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Taylor, J., Cunliffe, M. Multi-year assessment of coastal planktonic fungi reveals environmental drivers of diversity and abundance. ISME J 10, 2118–2128 (2016). https://doi.org/10.1038/ismej.2016.24
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2016.24
This article is cited by
-
Fungal parasitism on diatoms alters formation and bio–physical properties of sinking aggregates
Communications Biology (2023)
-
Impact of environmental factors on diversity of fungi in sediments from the Shenzhen River Estuary
Archives of Microbiology (2023)
-
Abundant and Rare Taxa of Planktonic Fungal Community Exhibit Distinct Assembly Patterns Along Coastal Eutrophication Gradient
Microbial Ecology (2023)
-
Intercomparison of Two Fluorescent Dyes to Visualize Parasitic Fungi (Chytridiomycota) on Phytoplankton
Microbial Ecology (2023)
-
Global contribution of pelagic fungi to protein degradation in the ocean
Microbiome (2022)
