
Abstract
Identifying soil microbial feedbacks to increasing temperatures and moisture alterations is critical for predicting how terrestrial ecosystems will respond to climate change. We performed a 5-year field experiment manipulating warming, watering and their combination in a semiarid temperate steppe in northern China. Warming stimulated the abundance of genes responsible for degrading recalcitrant soil organic matter (SOM) and reduced SOM content by 13%. Watering, and warming plus watering also increased the abundance of recalcitrant SOM catabolism pathways, but concurrently promoted plant growth and increased labile SOM content, which somewhat offset SOM loss. The treatments also increased microbial biomass, community complexity and metabolic potential for nitrogen and sulfur assimilation. Both microbial and plant community composition shifted with the treatment conditions, and the sample-to-sample compositional variations of the two communities (pairwise β-diversity distances) were significantly correlated. In particular, microbial community composition was substantially correlated with the dominant plant species (~0.54 Spearman correlation coefficient), much more than with measured soil indices, affirming a tight coupling between both biological communities. Collectively, our study revealed the direction and underlying mechanisms of microbial feedbacks to warming and suggested that semiarid regions of northern steppes could act as a net carbon source under increased temperatures, unless precipitation increases concurrently.
Similar content being viewed by others

Introduction
Human activities have raised the concentration of atmospheric greenhouse gases, causing a 0.85 °C increase in the global mean temperature since 1880 (IPCC, 2014). Climate warming has profound influences on biodiversity and ecosystem functioning, which in turn can result in feedbacks to climate warming (Luo et al., 2014; Steinauer et al., 2015). Feedbacks of soil ecosystems to climate change remain poorly understood, despite containing more carbon (C) in the form of soil organic matter (SOM) than both aboveground plant communities and atmosphere pools combined (Grosse et al., 2011; Scharlemann et al., 2014). Climate change could enhance microbial decomposition of SOM, primarily by promoting enzyme activities, and lead to positive feedback through the direct release of CO2 and/or CH4 to the atmosphere (Heimann and Reichstein, 2008; Mackelprang et al., 2011; Karhu et al., 2014; McCalley et al., 2014). Alternatively, soil microbial communities could possibly acclimate/adapt to climate warming, and thus have no or even negative feedback (Zhou et al., 2012). A net increase or decrease in terrestrial ecosystem C storage will ultimately depend on the balance between plant primary productivity, microbial C fixation, soil heterotrophic respiration and soil C stabilization over time, and is likely to vary between ecosystem types. However, it has been challenging to identify the direction of microbial feedbacks to climate change, primarily because the recalcitrant components of SOM and the microbial groups responsible for their degradation are diverse, and SOM decomposition rates are slow (Curtis et al., 2002; Handelsman et al., 2007).
Temperature and precipitation are perhaps the most important factors in determining biodiversity and ecosystem functioning in terrestrial ecosystems, and precipitation patterns are expected to shift with climate warming (Dore, 2005; Groisman et al., 2005). Although the feedback of soil microbial communities to climate warming was found to be positive in a relatively arid prairie ecosystem (~350 mm mean annual precipitation), a negative feedback was revealed in a comparatively moist prairie ecosystem (~900 mm mean annual precipitation; Nie et al., 2012; Zhou et al., 2012). Therefore, it is important to investigate microbial feedbacks in additional ecosystem types and under concurrent changes of precipitation and temperature. Changes in moisture and temperature conditions also affect aboveground plant communities (Yang et al., 2011), which govern the type and abundance of many organic substrates provided to soil microbial heterotrophs. Thus, changes in climate should also impact microbial communities indirectly through shifts in plant composition and productivity.
The semiarid steppe ecosystem in northern China is an important component of the Eurasian grassland biome (Christensen et al., 2004; Niu et al., 2008), where water is a key-limiting factor and soil temperature is low from later autumn to early spring, limiting microbial activity (Liu et al., 2009; Zhang et al., 2014). According to climate history, air temperatures adjacent to our study site have risen by ~2.4 °C in the past several decades (1953–2008), and summer precipitation is expected to increase in the future (Sun and Ding, 2010). Here, we conducted a long-term field experiment involving watering (W) and warming (T), mimicking increased precipitation and elevated temperature, respectively, and their combination (WT). We employed shotgun metagenomic sequencing, which can reveal the presence and relative abundance of genes responsible for SOM transformations in situ and at the whole-community level, of soil microbial communities sampled after 5 years of treatment and integrated the resulting sequence data with plant community properties and soil physicochemical characteristics in order to identify the direction and mechanism of soil microbial feedbacks to climate change. The biomass and species richness of plants were also monitored in order to assess the interplay between aboveground plant and belowground microbial communities (Zhang et al., 2014).
Given the dry conditions and cool autumn/winter temperatures that characterize this ecosystem, we hypothesized that watering and warming will stimulate microbial growth/biomass and also increase community diversity/complexity (hypothesis 1). Enhanced microbial activities will result in greater utilization of recalcitrant SOM substrates and soil nutrients (hypothesis 2), which will be relatable to whole-community gene abundances and the corresponding environmental indices. Furthermore, the potential synchrony in plant and microbial community assemblages under this experimental regime, attributable to existing environmental parameters, treatment conditions, plant–microbe relationships, can be assessed by comparing the sample-to-sample compositional variations of both groups (hypothesis 3).
Materials and methods
Experimental design in the field
This experiment started in 2005 and was part of a long-term field experiment conducted in a steppe ecosystem in the Inner Mongolia Autonomous Region of China. The study site and experimental design have been described previously (Niu et al., 2008; Liu et al., 2009; Yang et al., 2011), and only a brief description is provided here. The site was a typical temperate zone habitat, characterized by a semiarid continental monsoon climate. Annual mean temperature was 2.1 °C, with monthly mean temperature ranging from –17.5 °C in January to 18.9 °C in July. Annual mean precipitation was ~385.5 mm, with 80% occurring from June to September. Soil was chestnut soil (Chinese classification), corresponding to the Calcis-orthic Aridisol in the US Soil Taxonomy classification, with sand, silt and clay being 62.7%, 20.3% and 17.0%, respectively. Soil mean bulk density was ~1.31 g cm−3. This ecosystem was dominated by perennials, including Agropyron cristatum, Allium bidentatum, Artemisia frigida, Cleistogenes squarrosa, Potentilla acaulis and Stipa krylovii.
This experiment adopted a paired nested design, with watering and warming as the primary and secondary factor, respectively (Niu et al., 2008; Liu et al., 2009; Yang et al., 2011). There were three pairs of 10 m × 15 m plots, with one plot in each pair being randomly assigned to the watering treatment and the other to the control (see Supplementary Figure S1 for a graphical representation of the experimental design). In each watering plot, six sprinklers were arranged evenly in two rows, with a 5 m distance between two sprinklers. Each sprinkler covered a circular area with a 3 m diameter, and the six sprinklers covered the whole 10 m × 15 m plot. In July and August, 15 mm water was added weekly to the watering plots, and thus a total amount of 120 mm water (~30% of annually mean precipitation) was added each year. Within each 10 m × 15 m plot, four 3 m × 4 m subplots were randomly assigned to the warming and control subplots (each with two replicates). The warming subplots were heated continuously since 28 April 2005, using 165 cm × 15 cm MSR-2420 infrared radiators (Kalglo Electronics, Bethlehem, PA, USA) suspended 2.5 m above the ground, and resulted in a ~1.1 °C increase in soil temperature above control conditions. In the control subplot, one ‘dummy’ heater with the same size and shape as the infrared radiator was suspended 2.5 m high, mimicking the shading effect of the heater. For the watering treatment, the sprinkler was very small and its disturbance effect on the ecosystem was negligible, so we did not install a ‘dummy’ sprinkler in the control. Overall, there were six replicates for each of the four treatments.
Measurement of soil physiochemical indexes, plant and bacterial community indexes
On 22 August 2010, four random soil cores (10 cm deep, 3.5 cm diameter) were collected from each 3 m × 4 m subplot. All soil cores were taken no less than two inches away from a plant stalk, and the aboveground foliage of plant material was removed before sampling. The four soil cores were thoroughly mixed and passed through a 2 mm sieve to remove plant roots. Part of the composited soil samples was frozen for DNA extraction, whereas the remaining portion was used to measure SOM content, soil total nitrogen (N) content, NH4+-N and NO3−-N content, water content and pH. SOM and total N contents were quantified with the potassium dichromate-vitriol oxidization method and the Kjeldahl acid-digestion method, respectively (Bao, 2000). Soil NH4+-N and NO3−-N contents were determined on a FIAstar 5000 Analyzer (Foss Tecator, Hillerød, Denmark) after extraction of fresh soil with 1 mol l−1 KCl. Soil water content was determined as the weight loss after drying for 24 h at 105 °C. Soil pH was measured in 1:2.5 (W/V) suspensions of soil in distilled water. In addition, soil temperature at the depth of 10 cm was recorded with a CR1000 datalogger (Campbell Scientific, Logan, UT, USA) at 1 h intervals from 4 June 2005. And bacterial 16S rRNA gene abundance was quantified using real-time PCR, as described before (Zhang et al., 2013a, b).
All aboveground plants were harvested in two random 0.3 m × 0.5 m quadrats from each plot; the plants were sorted into species, and then oven-dried at 65 °C for 48 h and weighted. The total weight of all these plants was calculated as the aboveground plant biomass, and the total species number was counted to represent plant richness.
For each of the soil physicochemical indexes and plant and bacterial community indexes, two-way analysis of variance with block as a random factor was adopted to reveal the effect of warming and watering and their interaction. Multiple comparison was also conducted among the four treatments; in particular, the Least Significant Difference method and Tamhane’s T2 method was adopted for equal and unequal variances, respectively.
Shotgun metagenomic sequencing, sequence annotation and taxonomic analysis
Soil DNA was extracted with the MoBio PowerLyzer PowerSoil DNA isolation kit according to the manufacturer’s instructions. To obtain sufficient DNA for the shotgun metagenomic sequencing and to ensure adequate representation of soil, 4–6 replicates were conducted for each sample (0.25 g soil per replicate; 1.0–1.5 g of total soil used for extraction for each soil composite sample). The laboratory protocol followed the description in the Illumina Paired-End Prep kit protocol. DNA was sheared mechanically and size-selected to ~180-bp and gel purified. Sequencing was performed in the Illuminia Hiseq 2000 system at Shanghai Majorbio Bio-pharm Technology Co., Ltd for 23 samples (six per treatment); and there was one control sample for which the library preparation and sequencing methodologies were slightly different from the others, and its sequences were incomparable to other data sets.
Shotgun sequencing resulted in 12.0±4.5 million sequences (mean±one s.d.), or 2.4±0.9 Gbp of sequencing effort, per sample (average of 14 Gbp per treatment), and 8.6±2.3 million sequences passed quality control for downstream analysis (Supplementary Table S1). The sequences were quality trimmed and annotated (see the details in the Supplementary materials and methods), and further processed with the DESeq2 package (Love et al., 2014), in which the differentially-abundant metabolic pathways between the treatments were identified and the false discovery rate from multiple testing was accounted for using Benjamini–Hochberg correction (adjusted P-values; Benjamini and Hochberg, 1995). We specifically focused on the effect of each treatment on the abundances of genes for the catabolic processes of various C-complexes with different decomposability, ranging from the highly recalcitrant lipids and phenolics to the more labile monosaccharides, sugar acids and sugar alcohols. More attention was also given to the genes for N/sulfate acquisition, biosynthesis, and metabolism.
The relative abundance of various prokaryotic phyla was determined by aligning the sequences to Greengenes 16S ribosomal database (DeSantis et al., 2006; see the details in the Supplementary materials and methods). The relative abundance of fungi relative to prokaryotes (archaea and bacteria) was determined by aligning these reads to the Silva database (Quast et al., 2013). Bacteria were found to make up ~94% of ribosomal gene fragments, and thus we further calculated bacterial taxonomic richness by sampling equal number of sequences.
Estimation of community complexity and functional richness
Community complexity estimations were performed on composite metagenomes, where the sequences of five replicate samples of each treatment were combined (pooling of five samples in T, W and WT treatments to be consistent with the control group). All merged sequences ⩾80 bp were inputted to Nonpareil, which is a statistical tool that uses sequence redundancy to derive dataset complexity estimations (that is, total sequence complexity) and the amount of sequencing effort needed to achieve a desired representation of total sequence richness (Rodriguez-R and Konstantinidis, 2014).
To exclude the influence of unequal sampling, the relatively rarer functional pathways with <10−6 relative abundance in each sample (there were at least 106 reads assigned to functional genes for each sample) were removed for the calculation of pathway richness (Fierer et al., 2012).
Statistical analysis of microbial and plant compositional variation
Bray–Curtis and weighted UniFrac distances calculated from the relative abundances of OTUs (operational taxonomic unit), and Bray–Curtis distances calculated from the relative abundances of functional genes, were used to assess the compositional variation between microbial communities (Bray and Curtis, 1957; Lozupone et al., 2007). Bray–Curtis distances were also calculated from the biomass of plant species to represent plant community compositional variation of plots from which metagenomes were derived. Principal coordinate analysis was used to visualize the relative differences in community taxonomic and gene functional composition among different treatments (Anderson, 2003). PERMANOVA (permutational multivariate analysis of variance) was further used to reveal the effects of experimental treatments on the taxonomic and functional composition (Anderson, 2005). Linear regression analyses were used to identify the relationships between these principal coordinate analysis axes. Linear regression analyses were also used to identify the relationships between the relative change in phylum or functional gene abundance caused by different treatments.
Mass analysis between subsets of environmental indices and microbial community dissimilarity matrices
Mash, a tool that uses kmers to compare the sequence composition between metagenomes, was used to calculate sample–sample dissimilarities (options: -k 25 –s 100000; Ondov et al., 2015). The distance matrix obtained from Mash and from weighted UniFrac distances of 16S rRNA-derived OTUs were used to test for correlation between microbial community structure and biotic and abiotic variables with ‘bioenv’ (Clarke and Ainsworth, 1993), an analysis contained in the R package ‘vegan’. In short, this function calculates Euclidean distances between samples and attempts to determine the subset of environmental variables that correlates best to the user provided distance matrix. Subsets are scored and ranked with Spearman correlation. The Mash distances were also compared with plant community Bray–Curtis distances (see above) to determine whether the two distance matrices were significantly correlated using the Mantel test.
Accession numbers
The pyrosequencing reads were deposited in the Sequence Reads Archive database of the National Center for Biotechnology (accession no. SRA057669), and the shotgun metagenomic data were deposited in MG-RAST with the project name of ‘Multifactorial Environmental Changes in Inner Mongolia of China’.
Results and discussion
Microbial feedback direction under climate change
The warming treatment raised soil temperature at the sampling depth by 1.1 °C, on average, above the temperature of the control plots, which represented a small but significant shift in temperature relevant for climate change. Two-way analysis of variance revealed that warming and watering had significant interactive effect on the SOM content (P<0.05; Table 1). In particular, warming alone decreased the SOM content by ~13.0% relative to the control, but the decrease was smaller/negligible under watering (4.9%) and its combination with warming (3.8%; Table 1). Our shotgun sequencing covered ~30% of the total diversity (sequence richness) in each treatment (Supplementary Figure S2) based on estimates using Nonpareil (Rodriguez-R and Konstantinidis, 2014). Although this coverage may appear relatively low, the large number of replicated samples per treatment (5–6 replicates) and the fact that all data sets were similar in size, which makes comparisons among data sets robust (Rodriguez-R. and Konstantinidis, 2014), offset the low coverage and provided for meaningful comparisons.
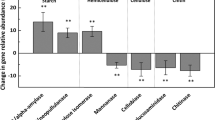
Watering, warming and their combination consistently increased the relative abundance of catabolic pathways specific to highly recalcitrant C-complexes, decreased those of medium decomposability, and had inconsistent effects on those of labile C-complexes (for example, poly-, di- and monosaccharides, sugar acids and alcohols) relative to the control samples. Specifically, genes involved in lignin degradation were 22.3% and 18.3% more abundant in W and WT treatments, respectively, compared with control (P<0.05; Figure 1). Observed differences in lipid catabolic pathways over control were +12.7%, +10.9% and +18.3%, in W, T and WT treatments, respectively (P<0.05; Figure 1). Genes for benzoate catabolism differed in abundance from control by +12.2%, +12.3% and +12.1% in W, T and WT treatments, respectively (P<0.10; Figure 1). In addition, described catabolic pathways specific to phenolic or carboxylic acids generally increased in abundance over control for all three treatments in most cases (Figure 1). In contrast, genes responsible for the degradation of complex carbohydrates, such as hemicellulose xylan (−13.5% in W, −14.0% in T, −11.7% in WT; P<0.01) and polysaccharide starch (−33.9% in W, −41.0% in T, −29.6% in WT; P<0.05; Figure 1), decreased in abundance for treatment groups relative to control.

Log2-fold differences in gene abundance between treatment (W, T, WT) and control soil samples for selected SOM degradation pathways. The raw count data underwent a variance-stabilizing transformation in DESeq2, which is used for logarithmically distributed count data with low mean values that tend to have high variance. Pathways denoted with an upward (increase relative to control) or downward (decrease relative to control) carat were significantly different (P<0.05; see key).
A recent study found that, among all biological/physicochemical indices (for example, pH and water content), gene abundance was the best predictor of soil element-cycling rates because it integrated the information of both environmental history and recent process activity (Petersen et al., 2012). Similarly, a study on soil gene–enzyme relationships demonstrated that, when accounting for existing microbial community structure and major environmental indices, gene relative abundance was an adequate predictor of the associated enzyme activity (Trivedi et al., 2016). Thus, although the increased abundance of pathways for recalcitrant SOM substrates does not represent a direct measure of enhanced microbial activity, these results suggest that microbes increased their usage of recalcitrant SOM substrates as a result of the experimental treatment conditions. This could be attributable to C compounds of medium decomposability becoming less abundant after 5 years of treatment conditions (Figure 1). Consistent with these interpretations, warming promoted microbial utilization of recalcitrant SOM such as phenols, measured by BIOLOG EcoPlates as described previously (Zhang et al., 2013c) and thus decreased the total SOM content (Table 1), with decreases in both the labile and recalcitrant fractions of SOM, which was quantified for soil samples taken at the same time (August 2010) from this field experimental system (Song et al., 2012). Meanwhile, warming had little effect on aboveground plant biomass (Table 1). Taken together, these results demonstrated that soil microbial communities of the Eurasian steppe under warming degraded more SOM, causing positive feedbacks to climate warming.
Experimental watering promoted an increase in plant richness (P<0.05, Table 1); this result provided a possible explanation for the difference in SOM content observed between warming and both watering groups; that is, an elevated diversity/input of plant organic materials into the soil, as reported previously (Song et al., 2012), partly counteracted the effect of microbial degradation and caused the change in the total SOM content to be smaller/negligible (Table 1).
Intensified assimilation of N and S into microbial biomass
Genes involved in the uptake and biosynthesis of N and sulfur (S) compounds were generally more abundant in treatment relative to control metagenomes (Figure 2). Specifically, sulfate binding (sbp) and import (cysAWT) genes were, on average, 13.5%, 9.7% and 7.8% more abundant in W, T and WT groups, respectively. Following S acquisition genes, pathways responsible for several steps involved in assimilatory sulfate reduction and incorporation of S into new biomass were also more abundant in treatment data sets. These genes included sulfate adenyltransferase (cysN, cysD, bifunctional nodQ), phosphoadenosine phosphosulfate reductase (cysH) and sulfite reductase (sir; Figure 2). Although genes related to nitrate and ammonium binding and acquisition showed mixed results, assimilatory nitrate reductase gene (nasA) was significantly more abundant by 27.5% and 26.6% in W and WT samples, respectively, relative to control (P<0.05, Figure 2). Although nitrate reductase genes napA and narG were more abundant in all treatment groups, genes specific to dissimilatory nitrate reduction (nrfA, nrfH) displayed mixed results, and genes specific to denitrification (nirK/S, norBC, nosZ) generally decreased in abundance across all treatment groups, relative to the control group. These results suggested that watering and warming treatments stimulated the growth of soil microorganisms and favored microbes capable of acquiring S and N inorganic nutrients. Consistent with these gene abundances, microbial biomass C and N were found to increase under these treatments, as reported previously (Zhou et al., 2013). In addition, the 16S rRNA gene abundance of soil bacterial communities based on quantitative PCR also showed an increase trend under the three treatments (19–26% increase), although it was non-significant statistically (P>0.10; Table 1).

A diagram representing selected nitrogen and sulfur pathways and the difference in abundance of the underlying genes between control and treatment metagenomes. Percentages are ordered like this: watering vs control, warming vs control and watering+warming vs control. Percentages given in blue or red represent an increase or decrease in the abundance of the corresponding genes, respectively. Percentages denoted with an asterisk (*) represent abundance changes that were statistically significant (P<0.05, DESeq2).
Stimulation of microbial community complexity and functional richness
Sequence-redundancy analysis using Nonpareil (Rodriguez-R and Konstantinidis, 2014) reveals that the composite metagenome of each treatment (W, T and WT) had greater overall sequence diversity than the composite control metagenome (Supplementary Figure S2), reflecting more complex microbial communities associated with these treatments. Thus, a larger sequencing effort is required to achieve a similar representation of the corresponding communities relative to the control one. For example, to reach 50% coverage, 13.4, 18.2, 15.7 and 16.6 billion base pairs of sequencing depth would be required for the control, watering, warming and watering, plus warming treatment, respectively. By combining five metagenomes for each sample group, ~30% of estimated community richness was represented in each case, with un-sampled diversity presumably belonging to sequences of comparatively rarer taxa.
Consistent with the result of community sequence complexity, there was greater microbial functional richness in the three treatment groups relative to the control, and this trend is especially obvious under watering, and watering plus warming (P<0.05; Table 1), which was congruent with the two treatments having the highest estimated community sequence complexity (see above). Both the relative abundance and species richness of bacteria was greater in the three treatments relative to the control (Table 1), and bacterial rRNA or functional genes made up >90% of the total, indicating that the stimulated community complexity and functional richness primarily resulted from bacteria rather archaea and fungi.
Warming and watering treatments likely resulted in more favorable metabolic conditions for soil microbes and thus increased the functional/metabolic diversity of their corresponding soil microbial communities. Several recent studies have demonstrated a positive correlation between gene diversity and ecosystem functioning (Salles et al., 2012; Singh et al., 2014; Trivedi et al., 2016); thus, the elevated complexity of these soil microbial communities indicated that the overall functional potential might have increased. Thus, it is likely that an overall increase in community diversity is at least partially responsible for the enhanced degradation of recalcitrant SOM in W, T and WT groups (Song et al., 2012).
High correlation between microbial composition and plant and soil indices
Experimental watering resulted in significant shifts in plant community composition (P<0.05). In particular, it primarily favored the growth of grasses and semi-shrubs compared with non-gramineous forbs, as reported previously (Yang et al., 2011). There was a significant positive correlation between plant community variation (that is, pairwise Bray–Curtis β-diversity distances) and the compositional variation of microbial communities (using Mash distances; Mantel test, R2=0.35, P<0.01; see also Supplementary Figure S3). These results imply that microbial communities with similar local plant communities tend to be more similar than those with different plant communities. The specific ecological mechanisms governing these patterns require additional investigation.
Furthermore, correlations were obtained between microbial community sample-to-sample similarity and environmental indices and plant biomass data (Clarke and Ainsworth, 1993). For distances based on kmer-composition of the metagenomes using Mash (Ondov et al., 2015), the maximum correlation obtained was 0.544 when using the abundance of six of the seven most abundant plant species (Supplementary Data S1). For distances derived from 16S rRNA OTUs (weighted unifrac), the maximum correlation achieved was 0.542, using three of the seven most abundant plant species, NH4+-N content, NO3−-N content and soil temperature (environmental indices included in assessment: Total C, Total N, NH4+-N, NO3−-N, soil moisture, pH, temperature, aboveground plant biomass, plant species richness). It is worth noting that distance matrices derived from sequence annotations, that is, Euclidean or Bray–Curtis distances derived from counts of reference proteins (for example, Swiss-Prot) or 16S-derived taxonomic summaries, which represent the more common practices, only provided weak correlations with plant and environmental indices (correlation coefficients <0.25). These results revealed that community composition was highly relatable to measured environmental and more so to plant parameters of the ecosystem, which is uncommon for soils owing to their high complexity and heterogeneity compared with other habitats, especially when sampling bulk soil (as opposed to the rhizosphere). It also demonstrates an improvement in associating microbial community composition with abiotic and biotic indices by incorporating sequence relatedness (weighted UniFrac) or sequence composition (Mash; kmer based) over commonly used functional gene annotation techniques. In previous studies, only a small part of microbial community variation could be explained by plant and soil indices, with the remaining fraction often attributed to under-sampling and ecological stochastic processes (Ramette and Tiedje, 2007; Ge et al., 2008; Wang et al., 2015).
Compared with the moderate-to-high correlation achieved between plant species community distances and Mash distances, a correlation of only 0.200 was observed when using solely abiotic environmental indices. This is likely because plant community composition is also reflective of local environmental conditions, as well as interactions between plants and belowground microbial communities, which might be un-relatable to soil conditions. This finding suggests that an alteration in environmental conditions directly affecting one biological community (for example, plants) would influence the other group (belowground microbes) indirectly (if not directly), owing to associations between biological communities. However, further investigations are required in order to obtain a more quantitative understanding of the direct and indirect associations between plant and soil microorganisms.
In agreement with the abovementioned results of the correlation analysis, PERMANOVA revealed that watering and warming significantly changed the taxonomic and functional structure of soil microbial communities, respectively (P<0.05; Figures 3a and b and Supplementary Figure S4), and the principal coordinate analysis axes of the taxonomic and functional composition showed significant linear relationships to each other (P<0.05; Figure 3c). Consistently, PERMANOVA also revealed that watering significantly altered the taxonomic composition of soil bacterial communities measured with 454 pyrosequencing targeting at 16S rRNA genes, as reported previously (Zhang et al., 2013b). Meanwhile, the treatments had similar effects on soil microbial communities because the relative changes in phyla/functional abundance caused by the three treatments were positively correlated (Supplementary Figure S5).

Effects of experimental treatments on the taxonomic (a) and functional (b) composition of soil microbial communities, and the relationship between the taxonomic and functional compositions (c). The proportion of variation explained by taxonomic PCoA axes 1 and 2 is 41.41% and 22.10%, respectively. The proportion of variation explained by functional PCoA axes 1 and 2 is 44.92% and 20.77%, respectively.
As stated above, besides plant species biomass, microbial community composition was also correlated with soil NH4+-N and NO3−-N content, indicating that soil N content may have had an important role in structuring the microbial community. Meanwhile, soil NH4+-N content decreased under these treatments (Table 1), whereas microbial genes for N assimilation (Figure 2) and the activities of N-acquisition enzymes of microbial communities (Zhou et al., 2013) increased under treatment conditions, consistent with an overall higher demand for N. These results suggest that the effect of plants on belowground microbial communities might be mediated, as least in part, by higher demand for N. Experimental methods aimed at uncovering the extent to which plants and belowground microbial communities (the biological groups primarily mediating C fixation and release from soils) respond synchronously vs independently to these types of environmental change should continue to be a focal point in future research endeavors.
Conclusions
The SOM content is as high as ~2.4 kg m−2 in the 0–10cm soil layer, representing ~1.2% of the total soil weight in this semiarid steppe ecosystem. Because most soil organic material is very complex and recalcitrant, total SOM content often seems to be very stable for long periods of time (Lützow et al., 2006; Mikutta et al., 2006), even under the influence of climate warming (Zhou et al., 2012). Here, we found a significant decrease in the SOM content caused by just 5 years of moderate climate warming of ~1.1 °C (Table 1), providing strong evidence of microbial positive feedback to climate warming. This is because warming (and also watering) enhanced microbial population size (more so taxa capable of degrading recalcitrant SOM compounds and with pathways involving the acquisition of inorganic N and S nutrients), and stimulated community diversity and complexity. Meanwhile, we find that a concurrent increase in precipitation will wholly or partially counteract soil microbial positive feedback. The close synchrony between plant and belowground communities across this experimental regime underscores the value and necessity of investigating potential co-responses and interactions between groups in future studies, even for bulk (non-rhizosphere) soil. It will also be important to assess the gene activity level (for example, metatranscriptomics) in addition to gene presence (for example, metagenomics), and to obtain more samples to test for seasonal and inter-annual dynamics in order for a more complete picture to emerge.
References
Anderson MJ . (2003) PCO: a FORTRAN computer program for principal coordinate analysis. Department of Statistics, University of Auckland: New Zealand.
Anderson MJ . (2005) PERMANOVA: a FORTRAN Computer Program for Permutational Multivariate Analysis of Variance. Department of Statistics, University of Auckland: New Zealand.
Bao SD . (2000) Soil and Agricultural Chemistry Analysis. China Agriculture Press: Beijing, China.
Benjamini Y, Hochberg Y . (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Statist Soc B 57: 289–300.
Bray JR, Curtis JT . (1957). An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr 27: 325–349.
Christensen L, Coughenour MB, Ellis JE, Chen Z . (2004). Vulnerability of the Asian typical steppe to grazing and climatic change. Climatic Change 63: 351–368.
Clarke KR, Ainsworth M . (1993). A method of linking multivariate community structure to environmental variables. Mar Ecol Prog Ser 92: 205–219.
Curtis TP, Sloan WT, Scannell JW . (2002). Estimating prokaryotic diversity and its limits. Proc Natl Acad Sci USA 99: 10494–10499.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al. (2006). Greengenes, a chimera-checked 16 S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072.
Dore MHI . (2005). Climatic change and changes in global precipitation patterns: what do we know? Environ Int 31: 1167–1181.
Fierer N, Leff JW, Adams BJ, Nielsen UN, Bates ST, Lauber CL et al. (2012). Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc Natl Acad Sci USA 109: 21390–21395.
Ge Y, He J, Zhu Y, Zhang J, Xu Z, Zhang L et al. (2008). Differences in soil bacterial diversity: driven by contemporary disturbances or historical contingencies? ISME J 2: 254–264.
Groisman PY, Knight RW, Easterling DR, Karl TR, Hegerl GC, Razuvaev VN . (2005). Trends in intense precipitation in the climate record. J Climate 18: 1326–1350.
Grosse G, Harden J, Turetsky M, McGuire AD, Camill P, Tarnocai C et al. (2011). Vulnerability of high-latitude soil organic carbon in North America to disturbance. J Geophys Res 116: G00K06.
Handelsman J, Tiedje J, Alvarez-Cohen L, Ashburner M, Cann IKO, DeLong EE et al. (2007) The New Science of Metagenomics: Revealing the Secrets of Our Microbial Planet. The National Academies Press: Washington, DC, USA.
Heimann M, Reichstein M . (2008). Terrestrial ecosystem carbon dynamics and climate feedbacks. Nature 451: 289–292.
Intergovernmental Panel on Climate Change (IPCC).. (2014) Climate Change 2014: Impacts, Adaptation, and Vulnerability. Cambridge University Press: Cambridge, UK.
Karhu K, Auffret MD, Dungait JA, Hopkins DW, Prosser JI, Singh BK et al. (2014). Temperature sensitivity of soil respiration rates enhanced by microbial community response. Nature 513: 81–84.
Liu W, Zhang Z, Wan S . (2009). Predominant role of water in regulating soil and microbial respiration and their responses to climate change in a semiarid grassland. Glob Change Biol 15: 184–195.
Love MI, Huber W, Anders S . (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550.
Lozupone C, Hamady M, Kelley ST, Knight R . (2007). Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73: 1576–1585.
Luo C, Rodriguez-R LM, Johnston ER, Wu L, Cheng L, Xue K et al. (2014). Soil microbial community responses to a decade of warming as revealed by comparative metagenomics. Appl Environ Microbiol 80: 1777–1786.
Lützow MV, Kögel-Knabner I, Ekschmitt K, Matzner E, Guggenberger G, Marschner B et al. (2006). Stabilization of organic matter in temperate soils: mechanisms and their relevance under different soil conditions – a review. Eur J Soil Sci 57: 426–445.
Mackelprang R, Waldrop MP, DeAngelis KM, David MM, Chavarria KL, Blazewicz SJ et al. (2011). Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480: 368–371.
McCalley CK, Woodcroft BJ, Hodgkins SB, Wehr RA, Kim E, Mondav R et al. (2014). Methane dynamics regulated by microbial community response to permafrost thaw. Nature 514: 478–481.
Mikutta R, Kleber M, Torn MS, Jahn R . (2006). Stabilization of soil organic matter: association with minerals or chemical recalcitrance? Biogeochemistry 77: 25–56.
Nie M, Pendall E, Bell C, Gasch CK, Raut S, Tamang S et al. (2012). Positive climate feedbacks of soil microbial communities in a semiarid grassland. Ecol Lett 16: 234–241.
Niu S, Wu M, Han Y, Xia J, Li L, Wan S . (2008). Water-mediated responses of ecosystem C fluxes to climatic change in a temperate steppe. New Phytol 177: 209–219.
Ondov BD, Treangen TJ, Mallonee AB, Bergman NH, Koren S, Phillippy AM . (2015). Fast genome and metagenome distance estimation using MinHash. bioRxiv doi:http://dx.doi.org/10.1101/029827.
Petersen DG, Blazewicz SJ, Firestone M, Herman DJ, Turetsky M, Waldrop M . (2012). Abundance of microbial genes associated with nitrogen cycling as indices of biogeochemical process rates across a vegetation gradient in Alaska. Environ Microbiol 14: 993–1008.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl Acids Res 41: D590–D596.
Ramette A, Tiedje JM . (2007). Multiscale responses of microbial life to spatial distance and environmental heterogeneity in a patchy ecosystem. Proc Natl Acad Sci USA 104: 2761–2766.
Rodriguez-R LM, Konstantinidis KT . (2014). Estimating coverage in metagenomic data sets and why it matters. ISME J 8: 2349–2351.
Salles JF, Le Roux X, Poly F . (2012). Relating phylogenetic and functional diversity among denitrifiers and quantifying their capacity to predict community functioning. Front Microbiol 3: 209.
Scharlemann JPW, Tanner EVJ, Hiederer R, Kapos V . (2014). Global soil carbon: understanding and managing the largest terrestrial carbon pool. Carbon Manage 5: 81–91.
Singh BK, Quince C, Macdonald CA, Khachane A, Thomas N, Al-Soud WA et al. (2014). Loss of microbial diversity in soils is coincident with reductions in some specialized functions. Environ Microbiol 16: 2408–2420.
Song B, Niu S, Zhang Z, Yang H, Li L, Wan S . (2012). Light and heavy fractions of soil organic matter in response to climate warming and increased precipitation in a temperate steppe. PLoS One 7: e33217.
Steinauer K, Tilman D, Wragg PD, Cesarz S, Cowles JM, Pritsch K et al. (2015). Plant diversity effects on soil microbial functions and enzymes are stronger than warming in a grassland experiment. Ecology 96: 99–112.
Sun Y, Ding Y . (2010). A projection of future changes in summer precipitation and monsoon in East Asia. Sci China Earth Sci 53: 284–300.
Trivedi P, Delgado-Baquerizo M, Trivedi C, Hu H, Anderson IC, Jeffries TC et al. (2016). Microbial regulation of the soil carbon cycle: evidence from gene–enzyme relationships. ISME J 10: 2593–2604.
Wang X, Van Nostrand JD, Deng Y, Lü X, Wang C, Zhou J et al. (2015). Scale-dependent effects of climate and geographic distance on bacterial diversity patterns across northern China’s grasslands. FEMS Microbiol Ecol 91: doi: 10.1093/femsec/fiv133.
Yang H, Wu M, Liu W, Zhang Z, Zhang N, Wan S . (2011). Community structure and composition in response to climate change in a temperate steppe. Glob Change Biol 17: 452–465.
Zhang N, Liu W, Yang H, Yu X, Gutknecht JLM, Zhang Z et al. (2013c). Soil microbial responses to warming and increased precipitation and their implications for ecosystem C cycling. Oecologia 173: 1125–1142.
Zhang X, Barberán A, Zhu X, Zhang G, Han X . (2014). Water content differences have stronger effects than plant functional groups on soil bacteria in a steppe ecosystem. PLoS One 9: e115798.
Zhang X, Liu W, Schloter M, Zhang G, Chen Q, Huang J et al. (2013a). Response of the abundance of key soil microbial nitrogen-cycling genes to multi-factorial global changes. PLoS One 8: e76500.
Zhang X, Zhang G, Chen Q, Han X . (2013b). Soil bacterial communities respond to climate changes in a temperate steppe. PLoS One 8: e78616.
Zhou J, Xue K, Xie J, Deng Y, Wu L, Cheng X et al. (2012). Microbial mediation of carbon cycle feedbacks to climate warming. Nature Clim Change 2: 106–110.
Zhou X, Chen C, Wang Y, Xu Z, Han H, Li L et al. (2013). Warming and increased precipitation have differential effects on soil extracellular enzyme activities in a temperate grassland. Sci Total Environ 444: 552–558.
Acknowledgements
We thank professors Shiqiang Wan, Quansheng Chen, Wenming Bai and many others for setting up the experiment; Yi Ren, Qiuping Hu and many others in Shanghai Majorbio Bio-pharm Technology Co., Ltd. for help in sequencing. This research was supported by the Strategic Priority Research Program of CAS (XDB15010404) of China, and by the US Department of Energy (award DE-SC0004601). We also thank three anonymous reviewers for helpful suggestions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Zhang, X., Johnston, E., Li, L. et al. Experimental warming reveals positive feedbacks to climate change in the Eurasian Steppe. ISME J 11, 885–895 (2017). https://doi.org/10.1038/ismej.2016.180
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2016.180
This article is cited by
-
Nitrogen deposition experiment mimicked with NH4NO3 overestimates the effect on soil microbial community composition and functional potential in the Eurasian steppe
Environmental Microbiome (2022)
-
Assessment of microbial α-diversity in one meter squared topsoil
Soil Ecology Letters (2022)
-
Specific patterns and drivers of the bacterial communities in the sediment of two typical integrated multitrophic aquaculture systems
Aquaculture International (2022)
-
Population viability analysis of the endangered Dupont’s Lark Chersophilus duponti in Spain
Scientific Reports (2021)
-
Aggregate sizes regulate the microbial community patterns in sandy soil profile
Soil Ecology Letters (2021)
