
Abstract
The gastrointestinal (GI) ecosystem is increasingly understood to be a fundamental component of health, and has been identified as a new focal point for diagnosing, correcting and preventing countless disorders. Shotgun DNA sequencing has emerged as the dominant technology for determining the genetic and microbial composition of the gut microbiota. This technology has linked microbiota dysbioses to numerous GI diseases including inflammatory bowel disease, obesity and allergy, and to non-GI diseases like autism and depression. The importance of establishing causality in the deterioration of the host–microbiota relationship is well appreciated; however, discovery of candidate molecules and pathways that underlie mechanisms remains a major challenge. Targeted approaches, transcriptional assays, cytokine panels and imaging analyses, applied to animals, have yielded important insight into host responses to the microbiota. However, non-invasive, hypothesis-independent means of measuring host responses in humans are necessary to keep pace with similarly unbiased sequencing efforts that monitor microbes. Mass spectrometry-based proteomics has served this purpose in many other fields, but stool proteins exist in such diversity and dynamic range as to overwhelm conventional proteomics technologies. Focused analysis of host protein secretion into the gut lumen and monitoring proteome-level dynamics in stool provides a tractable route toward non-invasively evaluating dietary, microbial, surgical or pharmacological intervention efficacies. This review is intended to guide GI biologists and clinicians through the methods currently used to elucidate host responses in the gut, with a specific focus on mass spectrometry-based shotgun proteomics applied to the study of host protein dynamics within the GI ecosystem.
Similar content being viewed by others


Introduction
Humans and our microbial inhabitants have co-evolved to form an aggregate organism. In the distal gastrointestinal (GI) tract, where microbes are at the greatest abundance, the complex interactions between host and microbes define health and disease. This dynamic relationship differs between individuals (Costello et al., 2009) and changes over time due to numerous variables including diet (Turnbaughet al., 2009), environment (Spor et al., 2011) and host genetics (Turnbaugh et al., 2009). Intensive effort has been applied toward characterizing the microbiota composition and functionality. The MetaHIT consortium (Qin et al., 2010) and the Human Microbiome Project (Consorium THMP, 2012) among other initiatives have charted the diversity of the gut microbiota across the modern world. Elegant work using mouse models has established mechanisms underlying complex host–microbiota interactions (Ivanov et al., 2009; Hsiao et al., 2013; Smith et al., 2013), although relatively few studies have been extended to include mechanistic connections to human biology (Nicholson et al., 2005; Koeth et al., 2013). Potentially significant differences in the microbiota between animal models and humans, co-evolved interactions between a microbiota and its resident species (Snel et al., 1995; Ley et al., 2008) and difficulties in monitoring host responses in humans non-invasively have resulted in a major gap in our understanding of human–microbiota interdependencies. To complement the wealth of sequencing-based microbiota characterization, discovery-based efforts are necessary to measure the numerous ways hosts respond to their resident microbiota and identify novel proteins and pathways that mediate interaction (Box 1).
Current methods of host response analysis
Most analyses of host responses to the microbiota have been conducted with targeted, single-protein level molecular approaches. Although methods, including western blotting, ELISA and quantitative PCR, reliably validate functions and interactions of candidate molecules, they must be individually tailored to specific hypotheses, and are thus inherently low-throughput and narrow in scope. In an extremely complex, ill-defined system like the human gut, targeting only a few molecules for measurement is likely to overlook key players. Discovering physiological responses in an untargeted fashion is necessary for generating new hypotheses in a manner commensurate with ongoing, global, microbiota sequencing efforts (Table 1).
Large-scale transcriptional assays (RNAseq, microarray) of intestinal tissue provide a highly sensitive analysis of microbiota-influenced gene expression that has been applied in both human and model organism contexts (Laukens et al., 2006; Athanasiadou et al., 2011; El Aidy et al., 2012). Although the sensitivity of sequencing is unmatched in the '-omics' space, the vast constellation of cell types in the gut could confound these results by obscuring the contribution of ecologically important but low-abundant cells. An ideal assay would independently measure the contribution of each cell type from a single biopsy (Box 1). For example, goblet cells have an essential role in mucus production, which creates a barrier between the host and microbiota, but these cells only represent 4–16% of the intestinal epithelium (Kim and Ho, 2010). Laser-capture microdissection (Cash et al., 2006) and flow cytometry (Habib et al., 2012) have provided partial solutions for this problem by isolating cell types of interest. However, laser-capture microdissection is technically challenging and flow cytometry only separates cell types that can be unambiguously labeled and requires tissue disaggregation. Cell type-specific transcriptional assays are subject to the same sample limitations common to all transcript-level measurements: they are generally crude indicators of protein abundance (Gygi et al., 1999; Ideker et al., 2001) and cannot describe protein localization, chemical modifications or functional interactions. In the GI tract most direct host–microbe interactions are mediated by host proteins that are either secreted or present on the apical epithelial surface, and may represent a small signal that would be obscured by less relevant readouts of abundant intracellular transcripts. Furthermore, measuring host transcription requires tissue samples obtained through invasive procedures in humans, or sacrifice of animal models, thus establishing a substantial barrier to obtaining samples, particularly for the purpose of monitoring an individual's response to therapeutic interventions.
Imaging approaches have proven essential in differentiating disease states and severity. Histological sections were used to demonstrate the improvement of colitis in mice receiving daily administration of Faecalibacterium prausnitzii (Sokol et al., 2008) and in concordance with molecular approaches to describe the improvement in inflammation via treatment with acetate (Maslowski et al., 2009). Histology also provides spatial resolution to protein expression, both across the length of the gut and within a particular tissue. Conversely, the human requirement for pathology scoring increases variability. Furthermore, as imaging primarily measures cell or tissue morphological changes, it is less sensitive than molecular approaches and often requires many samples to achieve statistically meaningful results. Furthermore, biopsies are highly invasive, and only represent a small region of the tissue. More recently, in situ hybridization with species-specific, fluorescent probes (Swidsinski et al., 2005, 2007; Swidsinski and Sydora, 2007) have localized microbes within the intestinal space, adding an important dimension to unraveling the host–microbe relationship.
Metabolomics has shown great promise in the characterization of host–microbiota interactions. Metabolites in serum and feces, measured by one-dimensional nuclear magnetic resonance, provided the first glimpse of host–species interactions and temporal variation in the fecal metabolome (Saric et al., 2008). Similar studies correlated fecal metabolites with inflammatory bowel diseases (Le Gall et al., 2011) and associated fecal and urine metabolites with antibiotic treatments (Yap et al., 2008). Parallel approaches using mass spectrometry have correlated shifts in metabolic profiles measured from serum, urine and feces to underlying changes in the microbiota, but are unable to assign >95% of the features to known metabolites (Wikoff et al., 2009; Marcobal et al., 2013). Metabolomics demonstrates great promise for global GI studies. However, the absence of compound databases necessary to identify metabolites by either nuclear magnetic resonance or mass spectrometry and the difficulty to deconvolute host–microbiota co-metabolism are serious hurdles that need to be addressed.
As the focus of microbiome research in model organisms (and eventually humans) turns toward mechanistic experimentation and retrospective longitudinal assessments of large subject cohorts, non-invasive methods for assaying host health will be essential. Blood represents a commonly sampled biofluid from which host–microbe interactions can be measured. For example, serum cytokine profiles have suggested mechanisms by which gut microbes induce adaptive immune responses (Sokol et al., 2008). Cytokine measurements are sensitive and reliably represent systemic immune responses. However, as it is physically separated from the intestinal space and contacts many other body regions, serum cannot necessarily provide GI-specific immune profiles indicative of the current host–microbiota status. Furthermore, serum-based assays cannot quantify strictly gut-resident molecules, such as intestinal anti-microbial peptides, immunoglobulins, metabolic enzymes and mucus proteins.
Stool offers many advantages for measuring host responses to the microbiota within the GI tract. Importantly, stool is acquired non-invasively and contains molecules of both host and microbial origin from within the gut ecosystem. Where molecules identified from tissue biopsies and blood that can serve as proxies for microbiota–host interaction in the gut, molecules from stool can directly describe these interactions without confounding non-GI contributions. Moreover, as proteins in frozen fecal specimens are amenable to analysis, previously conducted microbiota-focused experiments can be revisited to provide additional host-specific insight.
The secretion of host proteins have a key role in the dynamics of host–microbial interactions (Vaishnava et al., 2011) and are conveniently measured from feces. Mass spectrometry-based proteomics is uniquely suited to discovering the proteins at the host–microbiota interface.
Proteomics in the gut
Proteomic studies of intestinal epithelial cells have characterized postnatal intestinal development (Hansson et al., 2011) and chronic inflammation (Shkoda et al., 2007). Unfortunately, these studies were subject to many of the same limitations as bulk transcriptomic analyses insofar as they had to surmount the noise of intracellular protein expression. Laser-capture microdissection of intestinal epithelial cells provides a limited amount of sample material, posing a significant constraint on the use of shotgun proteomic techniques. To identify and quantify the proteins secreted into the gut lumen, proteomic workflows had to adapt to the complexities of stool (Box 2).
In line with early proteomics approaches, the first studies of stool proteins relied on two-dimensional gel separation and spot excision (Klaassens et al., 2007). In 2009, the first global metaproteomic profile of human stool was conducted. Verberkmoes et al. (2009) used sequenced metagenomes from two previously surveyed individuals and 33 genomes of isolated commensal microbial species to enumerate candidate proteins in stool. Using this resource, they generated what, at the time, was the deepest coverage of a bacterial metaproteome (Table 2). By pelleting bacteria from fecal samples, bacterially associated host proteins and sloughed host epithelial cells contributed to the ~500 host-derived proteins they identified. In a subsequent study, these researchers improved their bacterial protein identification rates by using a matched metagenome–metaproteome approach: metagenomic data derived from individual fecal specimens were used to create matched protein sequence databases to aid in mass spectrometry-based protein identification (Erickson et al., 2012). More accurately, predicting the fecal proteome likely contributed to improved analytical sensitivity: >3000 bacterial and >1600 host proteins were identified across 12 individuals with Crohn’s disease. Through functional annotations and statistical analysis of host protein expression, they identified impaired epithelial integrity and decreased epithelial absorption as hallmarks of ileal Crohn's disease.
This matched metagenome–metaproteome approach is the current state of the art in whole-feces metaproteomics and addresses the issues of protein diversity and variability (Box 2). However, many challenges still exist. First, deep sequencing and de novo genome assembly are expensive, time consuming and technically challenging. Second, these approaches do not address dietary protein sources, which contribute to the GI ecosystem as well as to protein diversity. Third, proteomic data are reliant on the quality of genome annotations, which has yet to keep pace with the 10 million identified genes from the gut microbiota (Li et al., 2014). Finally, these metaproteomic approaches fall victim to the unavoidable issue of dynamic range (Box 2), even when considering recent advancements in mass spectrometry instrumentation. Although modern proteomic techniques are capable of measuring thousands of proteins from just hundreds of nanogram of input material (for example, see Hughes et al. (2014)), the competition from host, microbial and dietary proteins could decrease analytical sensitivity by orders of magnitude. Thus, it remains to be seen for which application proteomics of microbial proteins in the gut will contribute beyond the superior depth of RNAseq and metagenomics.
Proteomics as a tool for analyzing host responses
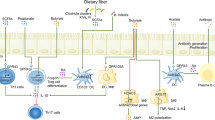
Host responses to the dense and dynamic intestinal ecosystem are often mediated by the secretion of proteins into the gut lumen (Vaishnava et al., 2011; Vaishnava and Hooper, 2007). Depleted of abundant intracellular proteins that often cloud metaproteomic studies, the extracellular component of the gut metaproteome offers a direct comparison of proteins produced by both the host and microbes that mediate their interactions. Importantly, none of the previously described host response analyses are capable of quantifying the relative abundance of these secreted host proteins in an untargeted and non-invasive way. We previously described a stool-based host-centric proteomics approach in which we identified and quantified thousands of host proteins present in the mouse intestinal lumen (Figure 1; Table 2). By removing intact cells (both microbial and host) and fecal debris, we were able to enrich for secreted proteins, many with well-characterized GI functions, such as digestion and immune response, and which serve as reporters of host physiological status in response to changing microbiota composition and function (Lichtman et al., 2013).

Gastrointestinal metaproteomics. Perturbations, like diet and infection, cause distinct changes to the host and microbiota. Protein-level changes throughout the gastrointestinal tract exist in the stool and can be assayed by mass spectrometry. Microbial-focused approaches pellet intact cells of both host and microbial origin, whereas the host-centric approach focuses on luminal proteins.
Many challenges remain in the analysis of host proteins in stool. The high concentration of molecules incompatible with sensitive liquid chromatography-mass spectrometry systems is an important issue to address for these methods to be widely accepted. Although the centrifugation strategy we previously described eliminates intact cells, sloughed epithelial cells that are broken down in the gut could still contribute intracellular proteins to the supernatant. Furthermore, identifying constitutive, stably expressed proteins in the gut will improve data normalization, much like is done with housekeeping genes in intracellular assays. Perhaps the greatest challenges are posed by clinical experiments, in which proteins will need to be associated with particular disease and microbiome states despite tremendous inter-individual variability in microbiota and overall stool composition. Normalization procedures that leverage stool-compatible quantitative labeling strategies (for example, reductive dimethylation (Hsu et al., 2003), tandem mass tags (Werner et al., 2014), Isobaric tag for relative and absolute quantitation (iTRAQ) (Ross et al., 2004) and stable isotope labeling in mammals (Wu et al., 2004)) or hypothesis-driven, targeted mass spectrometry analysis methods (for example, multiple reaction monitoring (Kennedy et al., 2014) and sequential window acquisition of all theoretical fragment ion spectra (SWATH-MS) (Gillet et al., 2012)) stand to improve upon the label-free quantification we previously used (Lichtman et al., 2013).
The application of this host-centric perspective has implications for basic biomedical research. Just as sequencing provided a powerful method to survey the microbial community, a similar approach is needed to reveal novel aspects of gut biology such as the discovery of pathways and effectors, unique biological states and signatures. Host-focused proteomics of stool promises to point the field toward proteins with yet-to-be-defined roles in governing interaction within the GI tract. The combination of microbial community, metabolite and host protein analyses from stool coupled with the application of gnotobiotic animal models and time-course studies will greatly improve our understanding of the proteins that mediate harmony within this complex ecosystem, and those that are signals of interactions going astray.
In clinical research, host-centric proteomics provides an orthogonal approach that could directly affect precision patient care. A multi-dimensional definition of GI states will allow for increased power in differentiating closely related but discrete states, the stratification of patients, individualized treatment and the monitoring of disease progression and recovery. These molecular phenotypes also provide a first step toward biomarker discovery. Signatures may be distilled to one or a few proteins that provide a simple means for diagnosing a spectrum of GI diseases. Such markers may be directly related to the underlying disease mechanism and direct pharmaceutical development, or they may serve as a proxy for specific biological events. As the stool is an aggregate read-out of the entire GI tract, biological insight need not be confined to the colon. Mapping protein signatures back to specific regions from which they originate promises clinical rewards. For example, enzymes produced in the pancreas have been assayed in stool as a metric for pancreatic function for more than 20 years (Loser et al., 1996; Lankisch, 1993).
In summary, we are now armed with high-throughput means to elucidate host intestinal states without invasive testing procedures. Host-centric proteomics is therefore compatible with long time-course evaluations of human subjects. These host-centric methods can be directly applied to banked stool samples from previous microbial studies, enriching prior data without the need for new specimen procurement. Although individualized microbiota present challenges in establishing microbial signatures as markers of disease, host responses are likely to be more conserved. Rapidly improving mass spectrometry instrumentation, advancements in de novo peptide sequencing and multiplexed quantification tools are diminishing the problems associated with proteome complexity and dynamic range and thus greatly improving our abilities to dive deeper into the gut proteome (Box 2). Although discovery-based mass spectrometry techniques will not likely be a clinical tool for diagnosing patients, the markers identified could be readily transferred into targeted mass spectrometry (Addona et al., 2009; Whiteaker et al., 2011) or ELISA assays that are readily adapted by clinical labs.
Protein diversity
Standard shotgun proteomic workflows compare experimental fragmentation spectra of individual peptides to hypothetical spectra calculated from a database of potential peptide matches. Analyzing human stool from thousands of individuals across the world, metagenomic sequencing efforts have assembled between five and nine million unique open reading frames, corresponding with ~1 billion unique tryptic peptides. This is over 200 times larger than the human proteome and results in increased computational time and greatly decreased ability to distinguish correct identifications from spurious matches. Single-nucleotide polymorphisms and post-translational modifications, though biologically relevant, further exacerbate the search space problem, and are best addressed through alternate means, including iterative database search procedures (Bern et al., 2012) and de novo sequencing (Ma et al., 2003).
Individual variability
Although the total sequenced metagenomic space is 5–9 million open reading frames, any single individual harbors only a subset of these genes. Accordingly, individual-to-individual variation in the microbiota is extensive (Costello et al., 2009). Paradoxically, considering all known metagenomic open reading frames would increase the frequency of false-positive identifications, while considering a more focused, though unmatched sequence database would not contain every protein in the sample. In the former case, database search engines are more likely to make an incorrect assignment when they are presented with more candidate peptide matches to an input spectrum—even when the true peptide is considered (Resing et al., 2004). In the latter, database search engines almost always return their 'best guess' as to the peptide source of a mass spectrum, even if the true source is missing from the sequence database. In both cases, higher numbers of false-positive identifications greatly decreases the ability to clearly discern which identifications are actually correct, and therefore decreases the sensitivity of the metaproteome analysis.
Dynamic range
If it can be assumed that the dynamic range of protein concentrations within a bacterium is on the order of 106 and the dynamic range of bacteria concentrations in the gut is, conservatively, on the order of 1010, then the total bacterial protein dynamic range is 1016. Compared with the quantitative dynamic range of 105 in shotgun proteomics, 1016 is a daunting challenge. Cell- and tissue-based shotgun proteomics analyses tend to use fractionation to extend dynamic range by separating abundant and less-abundant proteins but this has only been shown to improve the dynamic range slightly and comes at the cost of more analysis time on the mass spectrometer. This problem is evident in metaproteomic studies where only the most abundant (and well-characterized) bacterial proteins have been identified.
Solutions
Both matched metagenomics–metaproteomics and de novo peptide sequencing are viable solutions to the problems of diversity and variability but come with their own limitations. The dynamic range problem cannot be readily resolved with instrumentation or blind fractionation alone. The utilization of gnotobiotic animal models to decrease the microbial complexity and control the dynamic range may help, but the possibility of collecting deep proteomic signatures of the entire gut microbiota is not technically feasible, or possible in humans. The tried-and-true principles of proteomics are equally merit worthy in the metaproteome context: fractionation and enrichment. In the gut, this can include the rational enrichment of sub-proteomes, like those at the epithelial–luminal interface or the secreted proteome. Furthermore, these sub-proteomes may actually contain the proteins that actively define the host–microbiota relationship.
References
Addona Ta, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM et al. (2009). Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol 27: 633–641.
El Aidy S, van Baarlen P, Derrien M, Lindenbergh-Kortleve DJ, Hooiveld G, Levenez F et al. (2012). Temporal and spatial interplay of microbiota and intestinal mucosa drive establishment of immune homeostasis in conventionalized mice. Mucosal Immunol 5: 567–579.
Athanasiadou S, Jones La, Burgess STG, Kyriazakis I, Pemberton AD, Houdijk JGM et al. (2011). Genome-wide transcriptomic analysis of intestinal tissue to assess the impact of nutrition and a secondary nematode challenge in lactating rats. PLoS One 6: e20771.
Bern M, Kil YJ, Becker C . (2012). Byonic: advanced peptide and protein identification software. Curr Protoc Bioinformatics Chapter 13: Unit13.20. doi:10.1002/0471250953.bi1320s40.
Cash H, Whitham C, Behrendt C . (2006). Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313: 1126–1130.
Human Microbiome Project Consorium. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214.
Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R . (2009). Bacterial community variation in human body habitats across space and time. Science 326: 1694–1697.
Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C et al. (2012). Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One 7: e49138.
Le Gall G, Noor SO, Ridgway K, Scovell L, Jamieson C, Johnson IT et al. (2011). Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J Proteome Res 10: 4208–4218.
Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L et al. (2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 11: 016717.
Gygi SP, Rochon Y, Franza BR, Aebersold R . (1999). Correlation between protein and mRNA abundance in yeast correlation between protein and mRNA abundance in yeast. Mol Cell Biol 19: 1720–1730.
Habib AM, Richards P, Cairns LS, Rogers GJ, Bannon CAM, Parker HE et al. (2012). Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology 153: 3054–3065.
Hansson J, Panchaud A, Favre L, Bosco N, Mansourian R, Benyacoub J et al. (2011). Time-resolved quantitative proteome analysis of in vivo intestinal development. Mol Cell Proteomics 10: 005231.
Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T et al. (2013). Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155: 1451–1463.
Hsu JL, Huang SY, Chow NH, Chen SH . (2003). Stable-isotope dimethyl labeling for quantitative proteomics. Anal Chem 75: 6843–6852.
Hughes CS, Foehr S, Garfield Da, Furlong EE, Steinmetz LM, Krijgsveld J . (2014). Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol 10: 757–757.
Ideker T, Thorsson V, Ranish Ja, Christmas R, Buhler J, Eng JK et al. (2001). Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 292: 929–934.
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485–498.
Kennedy J, Abbatiello S, Kim K . (2014). Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat Methods 11: 149–155.
Kim YS, Ho SB . (2010). Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep 12: 319–330.
Klaassens ES, de Vos WM, Vaughan EE . (2007). Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract. Appl Environ Microbiol 73: 1388–1392.
Koeth Ra, Wang Z, Levison BS, Buffa Ja, Org E, Sheehy BT et al. (2013). Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19: 576–585.
Lankisch PG . (1993). Function tests in the diagnosis of chronic pancreatitis. Critical evaluation. Int J Pancreatol 14: 9–20.
Laukens D, Peeters H, Cruyssen BV, Boonefaes T, Elewaut D, De Keyser F et al. (2006). Altered gut transcriptome in spondyloarthropathy. Ann Rheum Dis 65: 1293–1300.
Ley R, Hamady M, Lozupone C, Turnbaugh P, Ramey RR, Bircher JS et al. (2008). Evolution of mammals and their gut microbes. Science 320: 1647–1652.
Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S et al. (2014). An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 32: 834–841.
Lichtman JS, Marcobal A, Sonnenburg JL, Elias JE . (2013). Host-centric proteomics of stool: a novel strategy focused on intestinal responses to the gut microbiota. Mol Cell Proteomics 12: 3310–3318.
Loser C, Mollgaard A, Folsch UR . (1996). Faecal elastase 1: a novel, highly sensitive, and specific tubeless pancreatic function test. Gut 39: 580–586.
Ma B, Zhang K, Hendrie C, Liang C, Li M, Doherty-Kirby A et al. (2003). PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom 17: 2337–2342.
Marcobal A, Kashyap PC, Nelson TA, Aronov PA, Donia MS, Spormann A et al. (2013). A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. ISME J 7: 1933–1943.
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D et al. (2009). Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461: 1282–1286.
Nicholson JK, Holmes E, Wilson ID . (2005). Gut microorganisms, mammalian metabolism and personalized health care. Nat Rev Microbiol 3: 431–438.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65.
Resing Ka, Meyer-Arendt K, Mendoza AM, Aveline-Wolf LD, Jonscher KR, Pierce KG et al. (2004). Improving reproducibility and sensitivity in identifying human proteins by shotgun proteomics. Anal Chem 76: 3556–3568.
Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S et al. (2004). Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 3: 1154–1169.
Saric J, Wang Y, Li J, Coen M, Utzinger J, Marchesi JR et al. (2008). Species variation in the fecal metabolome gives insight into differential gastrointestinal function. J Proteome Res 7: 352–360.
Shkoda A, Werner T, Daniel H, Gunckel M, Rogler G, Haller D . (2007). Differential protein expression profile in the intestinal epithelium from patients with inflammatory bowel disease. J Proteome Res 6: 1114–1125.
Smith P, Howitt M, Panikov N . (2013). The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 80: 569–574.
Snel J, Heinen PP, Blok HJ, Carman RJ, Allen PC . (1995). Comparison of 16 S rRNA sequences of segmented filamentous bacteria isolated from mice, rats, and chicketns and proposal of “Candidatus Arthromitus.”. Int J Syst Bacteriol 45: 780–782.
Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux J-J et al. (2008). Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA 105: 16731–16736.
Spor A, Koren O, Ley R . (2011). Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 9: 279–290.
Swidsinski A, Loening-Baucke V, Theissig F, Engelhardt H, Bengmark S, Koch S et al. (2007). Comparative study of the intestinal mucus barrier in normal and inflamed colon. Gut 56: 343–350.
Swidsinski A, Sydora B . (2007). Viscosity gradient within the mucus layer determines the mucosal barrier function and the spatial organization of the intestinal microbiota. Inflamm Bowel Dis 13: 963–970.
Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H . (2005). Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol 43: 3380–3389.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al. (2009). A core gut microbiome in obese and lean twins. Nature 457: 480–484.
Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI . (2009). The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1: 6ra14.
Vaishnava S, Hooper LV . (2007). Alkaline phosphatase: keeping the peace at the gut epithelial surface. Cell Host Microbe 2: 365–367.
Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O et al. (2011). The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 334: 255–258.
Verberkmoes NC, Russell AL, Shah M, Godzik A, Rosenquist M, Halfvarson J et al. (2009). Shotgun metaproteomics of the human distal gut microbiota. ISME J 3: 179–189.
Werner T, Sweetman G, Savitski MF, Mathieson T, Bantscheff M, Savitski MM . (2014). Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal Chem 86: 3594–3601.
Whiteaker JR, Lin C, Kennedy J, Hou L, Trute M, Sokal I et al. (2011). A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat Biotechnol 29: 625–634.
Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC et al. (2009). Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA 106: 3698–3703.
Wu CC, MacCoss MJ, Howell KE, Matthews DE, Yates JR . (2004). Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal Chem 76: 4951–4959.
Yap IKS, Li JV, Saric J, Martin F-P, Davies H, Wang Y et al. (2008). Metabonomic and microbiological analysis of the dynamic effect of vancomycin-induced gut microbiota modification in the mouse. J Proteome Res 7: 3718–3728.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Lichtman, J., Sonnenburg, J. & Elias, J. Monitoring host responses to the gut microbiota. ISME J 9, 1908–1915 (2015). https://doi.org/10.1038/ismej.2015.93
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.93
This article is cited by
-
A carbohydrate-active enzyme (CAZy) profile links successful metabolic specialization of Prevotella to its abundance in gut microbiota
Scientific Reports (2020)
-
MetaRibo-Seq measures translation in microbiomes
Nature Communications (2020)
-
MetaPro-IQ: a universal metaproteomic approach to studying human and mouse gut microbiota
Microbiome (2016)
-
Grain-rich diets altered the colonic fermentation and mucosa-associated bacterial communities and induced mucosal injuries in goats
Scientific Reports (2016)
-
The effect of microbial colonization on the host proteome varies by gastrointestinal location
The ISME Journal (2016)
