
Abstract
Bacteria play key roles in the ecology of both aquatic and terrestrial ecosystems; however, little is known about their diversity and biogeography, especially in the rare microbial biosphere of inland freshwater ecosystems. Here we investigated aspects of the community ecology and geographical distribution of abundant and rare bacterioplankton using high-throughput sequencing and examined the relative influence of local environmental variables and regional (spatial) factors on their geographical distribution patterns in 42 lakes and reservoirs across China. Our results showed that the geographical patterns of abundant and rare bacterial subcommunities were generally similar, and both of them showed a significant distance–decay relationship. This suggests that the rare bacterial biosphere is not a random assembly, as some authors have assumed, and that its distribution is most likely subject to the same ecological processes that control abundant taxa. However, we identified some differences between the abundant and rare groups as both groups of bacteria showed a significant positive relationship between sites occupancy and abundance, but the abundant bacteria exhibited a weaker distance–decay relationship than the rare bacteria. Our results implied that rare subcommunities were mostly governed by local environmental variables, whereas the abundant subcommunities were mainly affected by regional factors. In addition, both local and regional variables that were significantly related to the spatial variation of abundant bacterial community composition were different to those of rare ones, suggesting that abundant and rare bacteria may have discrepant ecological niches and may play different roles in natural ecosystems.
Similar content being viewed by others

Introduction
Bacterioplankton are a fundamental component of aquatic ecosystems, having an extremely high level of genetic diversity, and playing essential roles in global biogeochemical cycles (Newton et al., 2011). This means that understanding the diversity of aquatic bacteria and their biogeographical patterns is a major ecological goal, because it provides an insight into the deeper ecological processes and mechanisms that underlie and maintain bacterial diversity and ecosystem function (Hanson et al., 2012). However, unlike the extensive studies of biogeographical patterns of large animals and plants, works on the spatial scaling of bacterial diversity are still limited (Preston, 1948; Green and Bohannan, 2006; Martiny et al., 2006; Ramette and Tiedje, 2007; Logue and Lindström, 2008; Barberán and Casamayor, 2010; Jones et al., 2012; Brown et al., 2014). A further complicating fact is that most bacterial communities comprise a large number of species. A few of these species are very abundant, others are moderately abundant and a large number species, often called the ‘rare biosphere’, are represented by only a few individuals. This highly diverse and rare microbial biosphere is largely unexplored (Pedrós-Alió, 2012). Influential studies interpreted as showing that ‘everything is everywhere’ (developed by Baas-Becking, 1934) in microbial ecology have made use of the rare biosphere community as a way of testing large-scale biogeographical ideas. For example, using varied culture conditions to demonstrate the presence of protist taxa in a lake that was not ecologically suitable for them so that they were present at the site in such low numbers that they were undetectable by direct microscopy of uncultured samples (see, for example, Fenchel et al., 1997; Finlay, 1998; Hambright et al., 2015). In the past few years, a revolution in high-throughput and deep sequencing is allowing a much more direct identification of the rare biosphere community in an environment (Pedrós-Alió, 2012; Logares et al., 2014).
Recent high-throughput sequencing studies indicated that the ‘rare biosphere’ fraction of the bacterial community follows biogeographical patterns similar to those of the most abundant members of the community and have distinct spatial distribution patterns in Arctic Ocean and coastal Antarctic lakes (Galand et al., 2009; Logares et al., 2013). Other studies provided evidence of an occupancy–abundance relationship for soil and marine abundant bacteria communities (Nemergut et al., 2011), implying that the dispersal probability of rare bacteria is limited compared with abundant taxa (Holt et al., 2004; Logue, 2010). In this context, inland waters are particularly interesting as they have significantly more bacterial groups and are more diverse than marine waters on a global scale (Barberán and Casamayor, 2010). Until now, it has remained unclear whether rare bacterial taxa follow similar biogeographical patterns as abundant taxa in inland lake and reservoir ecosystems.
In general, microorganisms, like larger organisms, are influenced by complex and interacting sets of abiotic and biological processes, leading to variation in their community distribution patterns at different spatial scales (Green and Bohannan, 2006; Ramette and Tiedje, 2007; Logue and Lindström, 2008; Jones et al., 2012). For larger organisms it has been suggested that abundant taxa dominate major ecosystem processes (for example, carbon flow and nutrient cycling), whereas rare taxa can be regarded as propagule banks and play minor but nonnegligible roles but may switch to being more important abundant taxa as environmental conditions change (Grime, 1998; Magurran and Henderson, 2003). More recently, similar suggestions have been made for marine microbial systems (Pedrós-Alió, 2012; Logares et al., 2014), implying that abundant and rare bacterial taxa may have different ecological responses to environmental changes. In this study, we hypothesized that the possible controlling factors of biogeographical distribution of rare bacteria subcommunities were different from those of abundant bacteria subcommunities in lakes and reservoirs at a large scale.
Bacterial metacommunities are normally assembled by both local environmental variables (environmental filtering and biotic interactions) and regional factors (dispersal-related processes) (Fenchel, 2003; Nemergut et al., 2011; Liu et al., 2013). Some studies have showed that local factors are more important (Fierer and Jackson, 2006, Graham and Fine, 2008; Cavender-Bares et al., 2009; Wang et al., 2013), whereas other studies hold that bacterial communities are assembled by regional forces (Leibold et al., 2004; Cottenie, 2005; Martiny et al., 2006). Recently, Lindström and Langenheder (2012) depicted the relationship between regional and local factors along gradients of dispersal rate and selective strength. It has been suggested that the nature of bacterial communities is important in the creation of wider biogeographical patterns as the dispersal potential could affect the connection between community composition and local and regional factors (Lindström and Langenheder, 2012; Lear et al., 2014). The most abundant bacteria can disperse readily as there are, by definition, many more individuals that can potentially be involved in a dispersal event. In contrast, rare bacteria are less abundant, and hence their dispersal rate should be low compared with abundant taxa. Therefore, we hypothesized that the relative influence of regional and local factors for abundant bacterioplankton subcommunities were different from rare subcommunities.
In this study, we used high-throughput sequencing to investigate the aquatic bacterial community along a latitudinal gradient ranging from 24 to 50 °N (over 2700 km) in 42 lakes and reservoirs across China. We sort to determine and compare the biogeographical patterns and drivers for abundant and rare bacterial subcommunities at a continental scale. Specifically, we aimed to answer the following key questions: do abundant and rare taxa show similar or different biogeographical patterns in lakes and reservoirs at a continental scale? Are the controlling factors and their contribution to the geographical pattern of abundant bacterial subcommunities different from the rare ones?
Materials and methods
Study area and sampling
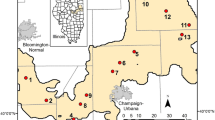
A total of 42 Chinese lakes and reservoirs, which were located between 24 and 50°N, were included in the sampling campaign (Figure 1 and Supplementary Table S1). Many of these lakes also featured in a study of aquatic testate amoebae by Ju et al. (2014). Field sampling took place from middle July to early August 2012. Surface water samples (upper 50–200 cm) in the epilimnion were collected at the center of each lake. All water samples were subsequently divided into two subsamples: one for water chemistry and the other for bacterial community analyses. All samples were stored in the dark at 4 °C and returned to the laboratory as soon as possible.

Location of the 42 sampling sites in China. FJ (included 5 reservoirs): Fujian Province, southeast China; CJ (9 lakes): the lower and middle reaches of Changjiang River, China; ECC (6 lakes): east central China, IM (13 lakes): Inner Mongolia, North China; NEC (9 lakes): northeast China.
Physicochemical analysis
Water temperature, electrical conductivity, pH, dissolved oxygen and turbidity of the epilimnion layer were measured in situ with YSI multi-parameter water quality sonde (YSI, Yellow Springs, OH, USA). Water depth of sampling site was measured with a Speedtech SM-5 Depthmate portable sounder (Speedtech Instruments, Great Falls, VA, USA). Water transparency was determined with a 30 cm Secchi disc. The concentrations of chlorophyll a, total nitrogen and total phosphorus were measured according to standard methods (Greenberg et al., 1992).
DNA extraction, PCR and high-throughput sequencing
A total of 500 ml water samples for bacterioplankton analyses were filtered through a 0.22-μm pore size polycarbonate filters (47 mm diameter, Millipore, Billerica, MA, USA) following Liu et al. (2013). The filters were stored at –80 °C until DNA extraction. Total DNA was extracted directly from the filter using FastDNA spin kit (Bio101, Carlsbad, CA, USA) according to the manufacturer’s instructions. Total DNA was sent to the Personal Biotechnology Co., Ltd. (Shanghai, China) for high-throughput sequencing on an Illumina MiSeq instrument (San Diego, CA, USA) using a paired-end 150-bp sequence read run. A set of primers was used to amplify the hypervariable V4 region (∼207 bp) of bacterial 16 S rRNA gene. In this study, the forward primer was 5′ -AYTGGGYDTAAAGNG- 3′, and the reverse primer was 5′ -TACNVGGGTATCTAATCC- 3′ (Claesson et al., 2009). Each DNA sample was individually PCR-amplified in triplicated 25 μl reactions included an initial denaturation at 94 °C for 5 min, followed by 25 cycles of 30 s at 94 °C, 30 s at 50 °C and 30 s at 72 °C. At the end of the amplification, the amplicons were subjected to final 7 min extension at 72 °C. Each reaction contained 1 × PCR buffer, 2.5 mM dNTPs, 0.625 U of Taq DNA polymerase, 10 μM of each primer and 20 ng of target DNA.
Sequence analysis
Raw sequence data were processed using MOTHUR v.1.33.3 (Schloss et al., 2009). Paired-end reads were merged. Sequences were then quality controlled with the flowing settings: any sequences length <150 or >300, average quality <30, ambiguous bases >0 and homopolymer length >6 were removed for further analysis. The remaining sequences were aligned to a reference alignment, and those sequences that did not align to the correct region were eliminated. To further reduce the noise in our sequences, we utilized pre-clustering and the resulting sequences were screened for chimeras using UCHIME (Edgar et al., 2011). We then used Bayesian classifier to classify those sequences against the Ribosomal Database Project 16S rRNA gene training set (version 9, http://rdp.cme.msu.edu). We required an 80% pseudobootstrap confidence score (Wang et al., 2007). All Archaea, Eukaryota, chloroplasts, mitochondria and unknown sequences were culled. Finally, sequences were split into groups according to their taxonomy and assigned to operational taxonomic units (OTUs) at a 3% dissimilarity level. The OTUs that contained <2 reads were not used to avoid possible biases. For our data analyses, we used a randomly selected subset of 26 322 sequences from each sample to standardize sequencing effort across samples.
In this study, the definition of abundant or rare OTUs combined their local and regional relative abundance. Locally abundant OTUs were defined as the OTUs with a representation of >1% within a sample, whereas locally rare OTUs were defined as having an abundance of <0.01% within a sample (Galand et al., 2009; Pedrós-Alió, 2012). We then calculated the average relative abundance of these locally abundant or rare OTUs across all samples. The OTUs that had a mean relative abundance of >0.1% were defined as regionally abundant OTUs, whereas the OTUs with a mean relative abundance of <0.001% were defined as regionally rare OTUs (Logares et al., 2014). The ecological literature usually considers ‘rarity’ to be a continuous variable – therefore, there is always a level of arbitrariness when defining a cutoff point for rarity in any given study (Gaston, 1994). To reduce this problem we have defined our ‘rare biosphere’ with reference to other recent publications, to facilitate comparisons between studies. In addition, to reduce the effect of arbitrary definition of abundant and rare OTUs, we performed multivariate cutoff level analysis to systematically estimate how our data sets are affected by the definition of abundant and rare OTUs (Gobet et al., 2010).
Data analyses
Bray–Curtis similarity matrix is considered to be one of the most robust similarity coefficients for ecological studies (Kent, 2012) and was applied to our community data set of bacterial OTU relative abundance. A nonmetric multidimensional scaling ordination was used to investigate differences in bacterioplankton communities between sites (Clarke and Gorley, 2001). Our study area includes several major climate types; thus, five regions were divided based on their climate and geographical characteristics (Ju et al., 2014). To evaluate the significant differences of bacterioplankton communities between these five regions, we used the randomization/permutation procedure analysis of similarities. The analysis of similarities statistic global R is calculated as the difference of between-group and within-group mean rank similarities, and thus it displays the degree of separation between groups. Complete separation is indicted by R=1, whereas R=0 suggests no separation (Clarke and Gorley, 2001). RELATE calculates rank correlation among the entire, abundant and rare bacterial community similarity matrices by PRIMER 5.0, and it thus provides a significance test with the matching coefficient ρm, equivalent to the Mantel’s test (Clarke and Gorley, 2001). We referred to positive and negative correlation levels between 0.5 and 1 as strong relationships at P<0.01.
Spearman’s rank correlations were used to determine the relationships between the Bray–Curtis similarity of bacterial community and the geographical distance of lakes or reservoirs, and the relationship between the Euclidean distance of environmental variables and the geographical distance.
A set of regional variables were generated through the use of principal coordinates of neighbor matrices (PCNM) analysis (Borcard and Legendre, 2002; Legendre et al., 2008) based on the longitude and latitude coordinates of each sampling site. The normality of the physicochemical variables were checked using Shapiro–Wilk test and variables were log(x+1) transformed with the exception of pH to improve normality and homoscedasticity for multivariate statistical analyses. Canonical correspondence analysis (CCA) was performed to explore the relationships between bacterial communities and physicochemical and PCNM variables. This method was chosen because preliminary detrended correspondence analysis on bacterial community data revealed that the longest gradient lengths were longer than 3.0, indicating that the majority of species exhibited unimodal responses to the environmental variation (Lepš and Šmilauer, 2003). Before the CCA, we used a forward selection procedure to select local physicochemical variables and regional variables using the ‘ordiR2step’ function from vegan (Blanchet et al., 2008). All nonsignificant (P>0.05) variables were eliminated in further analyses.
To determine the relative contribution of local (in this study, environmental variables) and regional (in this study, spatial factors based on PCNM) variables to the distribution of bacterial communities, standard and partial Mantel tests were also performed (Legendre and Legendre, 2012). The similarity matrices of bacterial community composition were obtained using Bray–Curtis index. The local and regional matrices were obtained using Euclidean distances with significant variables. PCNM, CCA, detrended correspondence analysis, forward selection procedure and Mantel tests were performed in the R language environment. Rarefaction curves and richness estimate of Chao 1 and ACE (abundance-based coverage estimator) were calculated in Vegan with R software (R Development Core Team).
Accession number
All sequence data from this study have been deposited in the public National Center for Biotechnology Information (NCBI) database (http://www.ncbi.nlm.nih.gov/) under the accession number SRX525963.
Results
The estimate of species richness and multivariate abundance cutoff
In total, 10 559 bacterial OTUs with 1 105 524 sequences were obtained in this study, and the number of bacterial OTUs varied from 816 (Donghaizi Lake) to 2026 (Dongxintunnanpao Lake) per sample (mean=1399, s.e.=48, n=42) (Supplementary Table S1). The total number of bacterial OTUs (10 559) was roughly equivalent to the number estimated by abundance-based richness estimators such as Chao 1 (10 791±22) and ACE (10 952±48). Both the estimated species-accumulation curves (Supplementary Figure S1) and extrapolated species richness indices (Chao 1 and ACE) (Supplementary Table S2) indicated that the majority of the pelagic bacterioplankton taxa had been recovered from the studied lakes and reservoirs.
At local and regional levels, 143 (1.4%) OTUs with 751 588 sequences (68.0%) were classified as abundant OTUs, whereas 7598 (72.0%) taxa and 29 824 (2.7%) sequences were rare OTUs (Supplementary Table S2). At local level, however, no OTU was always abundant (>1%) in all samples, and only two OTUs (including 143 102 sequences) with >1% abundance were present in >70% of the samples. In addition, 5435 OTUs (including 16 623 sequences) were always locally rare (<0.01%) in all samples (Supplementary Figure S2).
The multivariate cutoff level analysis showed that when the structure of community data were compared between the truncated and the original matrices, little variation in data structure was observed up to a removal of 45% of the rare part of the data set. On the other hand, when the increasing amount of rare types was >5%, the data structure of truncated matrices showed a little variation (Supplementary Figure S3). To fully capture all bacteria sequences in any environment is still impossible; however, our definitions of abundant (32.0%) and rare (2.7%) bacteria are reasonable and objective within the constraints of current technology.
Geographical patterns of bacterial community
We found a distinct biogeographical distribution pattern for the rare bacterial taxa that showed striking similarities with the geographical patterns of both abundant subcommunity (ρm=0.762, P<0.01) and entire community (ρm=0.807, P<0.01) (Figure 2). Interestingly, our five geographical regions were significantly separated at P=0.001 for the bacterioplankton communities, and the global R among the five groups was 0.398, 0.372 and 0.464 for the entire bacterial community, the abundant bacterial subcommunity and the rare bacterial subcommunity, respectively.

Multidimensional scaling (MDS) ordination for bacterioplankton communities from 42 lakes and reservoirs of China. All: all bacterial taxa; abundant: abundant taxa; and rare: rare taxa. For region abbreviations see Figure 1.
Distance effects on community composition and environmental variables
Overall, the similarity in bacterial community composition between any two lakes or reservoirs decreased with the increasing of geographic distance (Figure 3). For example, Spearman’s correlation analysis gave a correlation coefficient of –0.498 (P<0.01) between the similarity of entire bacterial community and geographic distance. In addition, Spearman’s correlation coefficients for abundant and rare taxa were –0.398 (P<0.01) and –0.507 (P<0.01) between the subcommunity similarity and geographic distance, respectively. In addition, lakes and reservoirs that were closer to each other presented more similar environmental conditions (Spearman’s correlation coefficient=0.137, P<0.01, Supplementary Figure S4).

Spearman’s rank correlations between the Bray–Curtis similarity of bacterioplankton community and geographical distance (n is the number of comparison).
Abundance–occupancy relationship
Bacterial relative abundance and local occupancy were positively correlated (r=0.409, P<0.01; Figure 4). In addition, 98% of abundant OTUs occupied >50% of sites, whereas no rare OTU occupied >50% of sites (Supplementary Figure S5).

Spearman’s rank correlation between median of bacterial OTU relative abundance and number of sites occupied (n is the number of OTUs).
Effects of local and regional factors on bacterioplankton distribution
The CCA ordination showed that four environmental variables (water temperature, electrical conductivity, turbidity and total nitrogen) and five regional factors (PCNM 1–3 and 13–14) were significantly related to the change of abundant bacterial subcommunity by forward model selection (P<0.05; Figure 5). However, the CCA for the rare bacterial subcommunity showed a different pattern. Three local environmental factors (electrical conductivity, transparency and depth) and six regional variables (PCNM 2–3, 6, 10 and 13–14) were significantly related to the variation of rare bacterial community composition (P<0.05; Figure 5).

CCA ordination showing the bacterial community composition in relation to significant local environmental variables and regional geographical factors (P<0.05). EC, electrical conductivity; TN, total nitrogen; Turb, turbidity; WT, water temperature.
The Mantel and partial Mantel results revealed that rare bacterial subcommunities were primarily governed by local environmental factors. However, the regional factors explained more variation in abundant bacterial distribution (Table 1).
Taxonomic distributions of abundant and rare bacteria
Actinobacteria (abundant 27.8±1.5% vs rare 7.3±0.6%, mean±s.e.), Bacteroidetes (14.0±1.5% vs 10.8±0.9%), Cyanobacteria (8.5±1.1% vs 2.3±0.2%), Firmicutes (0.4±0.2% vs 6.8±0.6%), Planctomycetes (2.8±0.5% vs 1.2±0.2%), Verrucomicrobia (21.2±1.9% vs 5.6±0.4%), Alpha-proteobacteria (5.9±0.4% vs 5.9±0.5%), Beta-proteobacteria (5.4±1.0% vs 4.1±0.4%) and Gamma-proteobacteria (4.1±0.5% vs 5.8±0.3%) taxa were the most frequent groups in both abundant and rare taxa. However, there were more unclassified bacterial groups (9.0±0.8% vs 39.0±1.1%) among the rare taxa, and a much higher number of taxonomic groups were present among rare bacteria compared with abundant bacteria (Supplementary Figure S6).
Discussion
Geographical patterns in abundant and rare bacteria
New molecular tools and increasing sampling effort have confirmed the existence of a ‘rare biosphere’ (Pedrós-Alió, 2012). The cosmopolitan theory proposes that rare phylotypes (species) are recruited through immigration (dispersal) and that they are protected from active loss by both viral lysis and predation because of their low abundance (Pedrós-Alió, 2006)—as in the long-standing ‘Everything is Everywhere’ approach to microbial biogeography that was developed mainly using morphospecies data (Finlay, 1998, 2002). This low loss rate runs counter to mid-twentieth century suggestions that the rarest planktonic organisms in a lake were likely to be most at risk of extinction (Hutchinson, 1964). This raises an important question because if rare microbes have a lower, or equal, extinction risk to common ones then this would be an area where the ecology of microbes differs from that of larger organisms where extinction risk is often associated with rarity (Caughley, 1994).
Recent studies using high-throughput sequencing in Arctic Ocean and coastal Antarctic lakes demonstrated that rare bacteria had distinct spatial distribution patterns, and showed a similar spatial pattern to the abundant bacterial subcommunity (Galand et al., 2009; Logares et al., 2013). Our results corroborated those previous findings, and provided further evidence from subtropical and temperate inland lakes and reservoirs to support the concept that rare bacteria exhibit biogeographical patterns. Furthermore, we found both abundant and rare bacterial communities strongly adhere to distance–decay relationship. This could be because of dispersal and/or because lakes close to each other tend to have similar environments. In general, these similar geographical distribution patterns in abundant and rare bacterial subcommunities suggested that the rare bacteria biosphere is not a random collection of taxa (Galand et al., 2009).
However, within this general pattern there were also some interesting differences between the distributions of abundant and rare bacteria. First, our results showed that abundant bacteria have a weaker distance–decay relationship (r=–0.398) than rare bacteria (r=–0.507, Figure 3). Second, the bacteria distribution frequency showed a significant positive relationship with the OTUs relative abundance in our studied lakes and reservoirs (Figure 4). These results indicate that the abundant bacteria with high local abundance have a decreased probability of local extinction and increase probability of dispersal, thereby resulting in a widespread or ubiquitous distribution. In fact, bacteria are of a size where long distance aerial dispersal is clearly possible (Wilkinson et al., 2012) and the more abundant taxa are presumably more likely to become airborne—perhaps via bubble burst processes associated with wind-created waves (Hamilton and Lenton, 1998). In addition, the commoner taxa are also more likely to be moved by waterbirds that have been shown to affect the movement of some zooplankton between lakes (Figuerola et al., 2005). An alternative, or additional, mechanism is that the abundant bacteria may grow on a wider spectrum of resources compared with the rare taxa, and hence leading to them being able to reach high population levels in a wider range of lakes (Hambright et al., 2015). Pedrós-Alió (2006) hypothesized that rare taxa have a cosmopolitan distribution, because of a low loss rate and a potentially unlimited dispersal capacity. Our results showed that the hypothesis that the rare biosphere has a cosmopolitan distribution did not apply in most cases. Conversely, our results suggest that abundant bacteria tend toward more cosmopolitan distributions, whereas the majority of rare bacteria are more likely to show restricted distributions. This matches the well-documented abundancy–occupancy relationship detailed for larger organisms (Gaston et al., 2000; Holt et al., 2004), suggesting similar ‘rules’ govern the ecology of microorganisms and the larger organisms that have more often been the subject of ecological study.
Controlling factors for geographical distributions of abundant and rare bacteria
An important ecological aspect for the understanding of biogeography is to determine the possible controlling factors and their contribution to spatial variation of bacterial community. Several local factors are known to affect bacteria species coexistence, including temperature (Liu et al., 2013), water chemistry (Berdjeb et al., 2011), quality and quantity of dissolved organic matter (Judd et al., 2006) and grazing (Pernthaler, 2005). However, spatial variation in the bacterial community is also influenced by regional factors such as dispersal limitation, mass effects and random demographics (Liu et al., 2013; Wang et al., 2013).
Our CCA analyses for abundant and rare bacterial subcommunities yielded different patterns because both communities were significantly related to different local and regional factors. A similar finding was reported by Kim et al. (2013) from an activated sludge bioreactor. They demonstrated that the temporal dynamics of abundant and rare bacterial taxa were constrained by different environmental variables. Hence, it is clear that abundant and rare bacterial taxa may have different ecological niches, and the majority of abundant and rare taxa may play different roles in our investigated lake and reservoir ecosystems. The few abundant taxa account for most of the biomass and carbon cycling, whereas some rare species may be important for the cycling of certain elements (Pedrós-Alió, 2012). An example from a peatland indicated that rare taxon Desulfosporosinus could contribute most of the sulfate reduction in the peat (Pester et al., 2010). Moreover, abundant and rare bacteria may have different ecological strategies. The resources that some abundant bacteria use may be more abundant and widespread; alternatively, some abundant bacteria, in general, may be able to grow on a wider spectrum of resources. In contrast, some bacteria may be adapted to use only a few specific substrates or to episodic situations of high nutrient abundance. For example, Teira et al. (2007) incubated natural bacterial assemblages in microcosms with different amendments of polycyclic aromatic hydrocarbons. During these experiments, when the polycyclic aromatic hydrocarbons were added, Cycloclasticus (a hydrocarbon-degrading gammaproteo bacterium) became an abundant member of the community. Once the polycyclic aromatic hydrocarbons had disappeared, the Cycloclasticus population reduced and returned to the rare biosphere. Therefore, rare microbial taxa can be regarded as a propagule bank (c.f., Grime, 1998). They may become an abundant member of the community, when the right environmental conditions are met or when the abundance of abundant microbes decreases drastically or even becomes extinct (Pedrós-Alió, 2012; Logares et al., 2014).
A major theme of ecological research is quantitatively characterizing the relative contribution of local versus regional factors. Partial Mantel tests provide a good solution to compare the relative importance of local and regional factors (Lindström and Langenheder, 2012). In this study, we found abundant bacterial subcommunities were mostly governed by regional factors, whereas the rare bacterial subcommunities were mainly structured by local environmental variables. Hanson et al. (2012) examined 19 previous studies and found that most studies suggested local factors had a greater effect on microbial composition than regional processes. More recently, Wang et al. (2013) illustrated that the dominance of local factors might occur when the selective strength of local habitat conditions is above a conceptual threshold. In the present study, our samples spanned a continental scale and comprised contrasting environmental conditions such as oligotrophic and eutrophic waters, different habitat types (for example, lakes vs reservoirs) and altitudes (Ju et al., 2014). However, the regional factors seem to be more important than local factors for the biogeographical pattern of abundant bacterial taxa in the lakes and reservoirs. This could be because some environmental variables that were related to spatial distribution of abundant bacterial subcommunity were not measured here. Another possible mechanism for this result is that the populations of abundant bacterial taxa exhibit massive immigration and this can result in more community variability being related to spatial variables that occur independently of any measured environmental variables (Lindström and Langenheder, 2012). Wang et al. (2013) examined the relative influence of deterministic (local factors) and stochastic (regional factors) processes for bacterial communities from various habitats. They suggested that stochastic processes may overwhelm deterministic process, when regional species pools were characterized by environmental generalists. Unsurprisingly, most abundant bacteria appeared to be habitat generalists in our study, given that 98% of abundant OTUs occupied >50% of sites. Similarly, Lear et al. (2014) found that the regional factors explained the largest proportion of variation of bacterial community composition in the ponds that are connected to each other by ephemeral channels. These authors suggested that large numbers of immigrant taxa are received by each pond that could enhance the connection between community composition and regional factors.
Conclusions
Our results demonstrated that the rare bacterioplankton subcommunity had a distinct biogeographical pattern in the studied Chinese lakes and reservoirs that was reasonably similar to that of the abundant bacteria. However, rare and abundant bacterial subcommunities were significantly related to different local and regional variables. Moreover, local processes and factors play the most important role in structuring rare bacterial assemblages, with regional factors explaining more variation in abundant bacterial distribution. In this study, we suggest that dispersal potentials and ecological roles of both rare and abundant bacteria are potentially different. Therefore, to obtain a comprehensive or full understanding and modeling of bacterial community biogeography, both groups should be distinguished in future studies.
References
Baas-Becking LGM. (1934) Geobiologie of Inleiding Tot de Milieukunde. WP Van Stockum & Zoon: The Hague: The Netherlands.
Barberán A, Casamayor EO . (2010). Global phylogenetic community structure and β-diversity patterns in surface bacterioplankton metacommunities. Aquat Microb Ecol 59: 1–10.
Berdjeb L, Ghiglione JF, Jacquet S . (2011). Bottom-up versus top-down control of hypo- and epilimnion free-living bacterial community structures in two neighboring freshwater lakes. Appl Environ Microbiol 77: 3591–3599.
Blanchet FG, Legendre P, Borcard D . (2008). Forward selection of explanatory variables. Ecology 89: 2623–2632.
Borcard D, Legendre P . (2002). All-scale spatial analysis of ecological data by means of principal coordinates of neighbor matrices. Ecol Model 153: 51–68.
Brown MV, Ostrowski M, Grzymski J, Lauro FM . (2014). A trait based perspective on the biogeography of common and abundant marine bacterioplankton clades. Mar Genom 15: 17–28.
Caughley G . (1994). Directions in conservation biology. J Animal Ecol 63: 215–244.
Cavender-Bares J, KozakK H, Fine PVA, Kembel SW . (2009). The merging of community ecology and phylogenetic biology. Ecol Lett 12: 693–715.
Claesson MJ, O’Sullivan O, Wang Q, Nikkila J, Marchesi JR, Smidt H et al. (2009). Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One 8: e6669.
Clarke KR, Gorley RN . (2001) PRIMER v5: User Manual/Tutorial. PRIMER-E: Plymouth: UK.
Cottenie K . (2005). Integrating environmental and spatial processes in ecological community dynamics. Ecol Lett 8: 1175–1182.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R . (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194–2200.
Fenchel T . (2003). Biogeography for bacteria. Science 301: 925–927.
Fenchel T, Esteban GF, Finlay BJ . (1997). Local versus global diversity of microorganisms: cryptic diversity of ciliate protozoa. Oikos 80: 220–225.
Fierer N, Jackson RB . (2006). The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA 103: 626–631.
Figuerola J, Green AJ, Michot TC . (2005). Invertebrate eggs can fly: evidence of waterfowl-mediated gene flow in aquatic invertebrates. Am Nat 165: 274–280.
Finlay BJ . (1998). The global diversity of protozoa and other small species. Int J Paristol 28: 29–48.
Finlay BJ . (2002). Global dispersal of free-living microbial eukaryote species. Science 296: 1061–1063.
Galand PE, Casamayor EO, Kirchman DL, Lovejoy C . (2009). Ecology of the rare microbial biosphere of the Arctic Ocean. Proc Natl Acad Sci USA 106: 22427–22432.
Gaston KJ . (1994) Rarity. Chapman & Hall: London, UK.
Gaston KJ, Blackburn TM, Greenwood JJD, Gregory RD, Quinn RM, Lawton JH . (2000). Abundance-occupancy relationships. J Appl Ecol 37: 39–59.
Gobet A, Quince C, Ramette A . (2010). Multivariate cut off level analysis (MultiCoLA) of large community datasets. Nucleic Acids Res 38: e155.
Graham CH, Fine PVA . (2008). Phylogenetic beta diversity: linking ecological and evolutionary processes across space in time. Ecol Lett 11: 1265–1277.
Green J, Bohannan BJM . (2006). Spatial scaling of microbial biodiversity. Trends Ecol Evol 21: 501–507.
Greenberg AE, Clesceri LS, Eaton AD . (1992) Standard Methods for the Examination of Water and Wastewater. American Public Health Association: Washington (DC), USA.
Grime JP . (1998). Benefits of plant diversity to ecosystems: intermediate, filter and founder effects. J Ecol 86: 902–910.
Hambright KD, Beyer JE, Easton JD, Zamor RM, Easton AC, Hallidayschult TC . (2015). The niche of an invasive marine microbe in a subtropical freshwater impoundment. ISME J 9: 256–264.
Hamilton WD, Lenton TM . (1998). Spora and Gaia: how microbes fly with their clouds. Etho Ecol Evol 10: 1–16.
Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JBH . (2012). Beyond biogeographic patterns: processes shaping the microbial landscape. Nat Rev Microbiol 10: 497–506.
Holt AR, Warren PH, Gaston KJ . (2004). The importance of habitat heterogeneity, biotic interactions and dispersal in abundance–occupancy relationships. J Anim Ecol 73: 841–851.
Hutchinson GE . (1964). The lacustrine microcosm reconsidered. Am Sci 52: 334–341.
Jones SE, Cadkin TA, Newton RJ, McMahon KD . (2012). Spatial and temporal scales of aquatic bacterial beta diversity. Front Microbiol 3: 1–10.
Ju LH, Yang J, Liu LM, Wilkinson DM . (2014). Diversity and distribution of freshwater testate amoebae (Protozoa) along latitudinal and trophic gradients in China. Microb Ecol 68: 657–670.
Judd KE, Crump BC, Kling GW . (2006). Variation in dissolved organic matter controls bacterial production and community composition. Ecology 87: 2068–2079.
Kent M . (2012) Vegetation Description and Data Analysis, 2nd edn. Wiley-Blackwell: Chichester, UK.
Kim T, Jeong J, Wells GF, Park H . (2013). General and rare bacterial taxa demonstrating different temporal dynamic patterns in an activated sludge bioreactor. Appl Microbiol Biotechnol 97: 1755–1765.
Lear G, Bellamy J, Case BS, Lee JE, Buckley HL . (2014). Fine-scale spatial patterns in bacterial community composition and function within freshwater ponds. ISME J 8: 1715–1726.
Legendre P, Bordard D, Peres-Neto P . (2008). Analyzing or explaining betadiversity? Comment. Ecology 89: 3238–3244.
Legendre P, Legendre L . (2012) Numerical Ecology, 3rd edn. Elsevier: Amsterdam: The Netherlands.
Leibold MA, Holyoak M, Mouquet N, Amarasekare P, Chase JM, Hoopes MF et al. (2004). The metacommunity concept: a frame work for multi-scale community ecology. Ecol Lett 7: 601–613.
Lepš J, Šmilauer P . (2003) Multivariate Analysis of Ecological Data Using CANOCO. Cambridge University Press: Cambridge, UK.
Lindström ES, Langenheder S . (2012). Local and regional factors influencing bacterial community assembly. Environ Microbiol Rep 4: 1–9.
Liu LM, Yang J, Yu XQ, Chen GJ, Yu Z . (2013). Patterns in the composition of microbial communities from a subtropical river: effects of environmental, spatial and temporal factors. PLoS One 8: e81232.
Logares R, Lindström ES, Langenheder S, Logue JB, Paterson H, Laybourn-Parry J et al. (2013). Biogeography of bacterial communities exposed to progressive long-term environmental change. ISME J 7: 937–948.
Logares R, Audic S, Bass D, Bittner L, Boutte C, Christen R et al. (2014). Patterns of rare and abundant marine microbial eukaryotes. Curr Biol 24: 813–821.
Logue JB, Lindström ES . (2008). Biogeography of bacterioplankton in inland waters. Freshw Rev 1: 99–114.
Logue JB . (2010) Factors influencing the biogeography of bacteria in freshwaters—a metacommunity approach. PhD thesis, Uppsala University. Uppsala: Sweden.
Magurran AE, Henderson PA . (2003). Explaining the excess of rare species in natural species abundance distributions. Nature 422: 714–716.
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4: 102–112.
Nemergut DR, Costello EK, Hamady M, Lozupone C, Jiang L, Schmidt SK et al. (2011). Global patterns in the biogeography of bacterial taxa. Environ Microbiol 13: 135–144.
Newton RJ, Jones SE, Eiler A, McMahon KD, Bertilsson S . (2011). A guide to the natural history of freshwater lake bacteria. Microbiol Mol Biol Rev 75: 14–49.
Pedrós-Alió C . (2006). Marine microbial diversity: can it be determined? Trends Microbiol 14: 257–263.
Pedrós-Alió C . (2012). The rare bacterial biosphere. Annu Rev Mar Sci 4: 449–466.
Pernthaler J . (2005). Predation on prokaryotes in the water column and its ecological implications. Nat Rev Microbiol 3: 537–546.
Pester M, Bittner N, Deevong P, Wagner M, Loy A . (2010). A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J 4: 1591–1602.
Preston FW . (1948). The commonness and rarity of species. Ecology 29: 254–283.
Ramette A, Tiedje JM . (2007). Biogeography: an emerging cornerstone for understanding prokaryotic diversity, ecology, and evolution. Microb Ecol 53: 197–207.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al. (2009). Introducing MOTHUR: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Teira E, Lekunberri I, Gasol JM, Nieto-Cid M, Álvarez-Salgado XA, Figueiras PG . (2007). Dynamics of the hydrocarbon-degrading Cycloclasticus bacteria during mesocosm-simulated oil spills. Environ Microbiol 9: 2551–2562.
Wang Q, Garrity GM, Tiedje JM, Cole JR . (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267.
Wang J, Shen J, Wu Y, Tu C, Soininen J, Stegen JC et al. (2013). Phylogenetic beta diversity in bacterial assemblages across ecosystems: deterministic versus stochastic processes. ISME J 7: 1310–1321.
Wilkinson DM, Koumoutsaris S, Mitchell EAD, Bey I . (2012). Modelling the effects of size on the aerial dispersal of microorganisms. J Biogeogr 39: 89–97.
Acknowledgements
We thank Zhengwen Liu, Kuanyi Li, Feizhou Chen and Bin Xue at Nanjing Institute of Geography and Limnology, Chinese Academy of Sciences, for field sampling assistance and providing part of physicochemical data. This work was funded by the National Basic Research Program of China (2012CB956103), the National Natural Science Foundation of China (31370471), the Natural Science Foundation for Distinguished Young Scholars of Fujian Province (2012J06009) and the Knowledge Innovation Program of the Chinese Academy of Sciences (IUEQN201305). We thank our referees for insightful comments that helped in improving the clarity of this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Liu, L., Yang, J., Yu, Z. et al. The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J 9, 2068–2077 (2015). https://doi.org/10.1038/ismej.2015.29
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.29
This article is cited by
-
Stochastic community assembly of abundant taxa maintains the relationship of soil biodiversity-multifunctionality under mercury stress
Soil Ecology Letters (2024)
-
Spatial distribution pattern across multiple microbial groups along an environmental stress gradient in tobacco soil
Annals of Microbiology (2023)
-
Core and conditionally rare taxa as indicators of agricultural drainage ditch and stream health and function
BMC Microbiology (2023)
-
Vegetation as a key driver of the distribution of microbial generalists that in turn shapes the overall microbial community structure in the low Arctic tundra
Environmental Microbiome (2023)
-
Assembly processes and functional diversity of marine protists and their rare biosphere
Environmental Microbiome (2023)
