
Abstract
Marine and estuary sediments contain a variety of uncultured archaea whose metabolic and ecological roles are unknown. De novo assembly and binning of high-throughput metagenomic sequences from the sulfate–methane transition zone in estuary sediments resulted in the reconstruction of three partial to near-complete (2.4–3.9 Mb) genomes belonging to a previously unrecognized archaeal group. Phylogenetic analyses of ribosomal RNA genes and ribosomal proteins revealed that this group is distinct from any previously characterized archaea. For this group, found in the White Oak River estuary, and previously registered in sedimentary samples, we propose the name ‘Thorarchaeota’. The Thorarchaeota appear to be capable of acetate production from the degradation of proteins. Interestingly, they also have elemental sulfur and thiosulfate reduction genes suggesting they have an important role in intermediate sulfur cycling. The reconstruction of these genomes from a deeply branched, widespread group expands our understanding of sediment biogeochemistry and the evolutionary history of Archaea.
Similar content being viewed by others


Introduction
Although a few cultured archaeal phyla provide most of our knowledge on archaeal metabolism, the uncultured majority of the archaeal domain remains essentially unaccounted for (Baker and Dick, 2013). Single-gene (for example, 16S ribosomal RNA (rRNA)) sequencing surveys have shown that uncultured archaea are extremely diverse in marine and estuary sediments (Durbin and Teske, 2011; Kubo et al., 2012). Stable carbon isotopic analyses of archaeal lipids and archaeal cells indicated that archaea assimilate buried organic carbon of photosynthetic origin in deep-sea sediments, suggesting a heterotrophic lifestyle (Biddle et al., 2006). Recently, partial genomes of two widespread sediment archaea groups (Marine Benthic Group D and Miscellaneous Crenarchaeaotal Group) revealed they are able to degrade detrital proteins as a source of carbon (Lloyd et al., 2013; Meng et al., 2014; Seyler et al., 2014). However with new uncultured archaeal groups being uncovered, new roles for archaea in carbon, nitrogen and sulfur cycling may also be discovered.
Estuaries are dynamic and productive environments that contain microbial communities essential to global nutrient cycling (Bauer et al., 2013). Microorganisms in estuary sediments facilitate the turnover of carbon and the anaerobic respiration of sulfur and nitrogen (Oremland and Polcin, 1982). Understanding the role of microorganisms in carbon cycling in sedimentary estuarine environments is globally important because estuaries provide a significant sink of atmospheric CO2 (Cai, 2011). In this study, we are focusing on the estuarine sediments and microbial communities of the White Oak River (WOR) in North Carolina, a typical blackwater river traversing coastal woodlands and swamps. The tidally influenced WOR estuary has served as a model system for geochemical and microbiological studies of anaerobic carbon cycling, especially methanogenesis and methane oxidation (Martens and Goldhaber, 1978; Kelley et al., 1990, 1995; Lloyd et al., 2011). Surveys of archaeal diversity of the WOR estuary sediment have revealed numerous uncultured archaea commonly seen in sediments throughout the world including Miscellaneous Crenarchaeotal Group, and Marine Benthic Groups B and D (Lloyd et al., 2011; Kubo et al., 2012; Lazar et al., 2014). The distinct redox zonation and high archaeal diversity of the WOR sediments make this site ideal for the environmentally contextualized reconstruction of novel genomes that will lend to better understanding of the metabolic and environmental potential of uncultured archaea.
De novo assembly and binning of random shotgun genomic libraries has proven to be an extremely powerful tool in reconstructing genomes of uncultured groups from the environment (Baker et al., 2010; Castelle et al., 2015) and determining their physiologies (Wrighton et al., 2012; Castelle et al., 2013, 2015; Hug et al., 2013). Advances in metagenomic assembly and binning techniques have enabled us to obtain genomes directly from the environment (Baker and Dick, 2013). In this study, we used this approach to reconstruct three unique, partial and near-complete genomes belonging to a deeply branched widespread and newly identified phylum of archaea, for which we propose the name ‘Thorarchaeota’; the reconstructed genomes provide a basis for inferring the evolutionary history and physiological potential of these sedimentary archaea.
Materials and methods
Sample collection and processing
Six 1-m plunger cores were collected from ~1.5 m water depth in three mid-estuary locations (two cores per site) of the White Oak River, North Carolina in October 2010 (site 1 at 34° 44.592′ N, 77° 07.435 ′ W; site 2 at 34° 44.482′ N, 77° 07.404′ W, and site 3 at 34° 44.141′ N, 77° 07298′ W). Cores were stored at 4 °C overnight and processed 24 h after sampling (Lazar et al., 2014). Each core was sectioned into 2-cm intervals. From each site one core was subsampled for geochemical analyses and the other core was subsampled for DNA extractions. A geochemical analysis of sulfate, sulfide and methane profiles of these cores indicated a distinct sulfur and methane-cycling zonation at each site. The sulfate-reducing zone started in sediment layers where sulfate was abundant and sulfide just started to accumulate (8–12 cm). The sulfate–methane transition zone (SMTZ) where porewater sulfate and methane overlapped, was located and sampled at around 26, 24 and 26 cm in sites 1, 2 and 3, respectively. The methane-producing zone was characterized by accumulating methane; a single sample from site 1 of this zone was taken at 52–54 cm (Lazar et al., 2014). DNA was extracted from these sediment depths using the UltraClean Mega Soil DNA Isolation Kit (MoBio, Carlsbad, CA, USA), using 6 g of sediment, and stored at −80 °C until use. A total of 100 ng of DNA was sheared to 270 bp using a focused ultrasonicator (Covaris, Woburn, MA, USA). The sheared DNA fragments were size selected using SPRI beads (Beckman Coulter, Brea, CA, USA). The selected fragments were then end-repaired, A-tailed and ligated of Illumina-compatible adapters (IDT, Inc., San Jose, CA, USA) using KAPA-Illumina library creation kit (KAPA Biosystems, Wilmington, MA, USA).
Genomic assembly, binning and annotation
Illumina (HiSeq) shotgun genomic reads were screened against Illumina artifacts (adapters and DNA spike-ins) with a sliding window with a kmer size of 28 and a step size of 1. Reads with 3 or more N’s or with average quality score of less than Q20 and a length <50 bps were removed. Screened reads were trimmed from both ends using a minimum quality cutoff of 5 using Sickle (https://github.com/najoshi/sickle). Trimmed, screened, paired-end Illumina reads were assembled using IDBA-UD (Peng et al., 2012) with the following parameters (—pre_correction —mink 55 —maxk 95 —step 10 —seed_kmer 55). To maximize assembly reads from different sites were coassembled.
The SMTZ assembly was generated from a combination of reads (698,574,240, average read length 143 bp and average insert 274 bp) from site 2 (30–32 cm) and 3 (24–28 cm). The methane-producing zone assembly was generated from high-quality reads (378,027,948, average read length 124 bp and average insert 284 bp) of site 1 (52–54 cm). As we were not able to coassemble all three of the SMTZ samples after running into computational limits at 1 TB RAM memory, one of the samples (site 1, 26–30 cm) was assembled separately from 345,710,832 reads (average length of 129 bp and average insert 281 bp). The contigs from this SMTZ sample were co-binned with the assembly of the other two SMTZ samples from sites 2 and 3, Contigs of genes of particular interest were checked for chimeras by looking for dips in coverage within read mappings.
Initial binning of the assembled fragments was done using tetra-nucleotide frequency signatures using 5 kb fragments of the contigs (Dick et al., 2009). The Emergent Self-organizing Maps were manually delineated and curated based on clusters within the map (as shown in Supplementary Figure S1). This binning was enhanced by incorporating coverage signatures for all of the assembled contigs into the Emergent Self-organizing Maps maps (Dick et al., 2009). Coverage was determined by recruiting reads (from each individual library/sample) to scaffolds by BLASTN (bitscore >75), which was then normalized to the number of reads from each library. Contigs from both assemblies were binned together, which resulted in the co-binning of SMTZ1-45 and SMTZ-45. These genomes were then separated based on their site of origin. The accuracy of the binning was then assessed by checking the genomic bins to which each of the 5 kb sub-portions of the contigs were assigned. If the contig was >15 kb then the contig was assigned to the bin that contained the majority of its 5 kb sub-portions. Some of the fragments were identified as contaminants based on differential coverage, GC content, phylogenetic placement and the presence of duplicate genes and were removed if necessary. Closely related variants of a lineage were retained in a bin. Binning was also manually curated based on GC content, top blast hits and mate pairings. The completeness, contamination and strain heterogeneity of the genomes within bins was then estimated by using CheckM (Parks et al., 2015).
Genes were called and putative function was assigned using the JGI IMG/MER system (Markowitz et al., 2012). The CAZy server (Marseille, France) (e-value cutoff of <1e−5) was used to identify all carbohydrate-active genes (Lombard et al., 2014); a subset of these have been shown to be involved in hydrolysis of extracellular carbohydrates (Wrighton et al., 2014). The full metagenomic assemblies presented in this study are available in IMG with the following IMG Taxon IDs: 3300002052 for the SMTZ site 1 assembly (‘SM1’ genomes) and 3300001753 for SMTZ sites 2 and 3 assembly (‘SM23’ genomes). The genomic bins supporting the results of this article are being made available in NCBI Genbank under the BioProjectID PRJNA270657. The genomes described in this study have been deposited in NCBI under BioProject PRJNA270657—SAMN03998758, SAMN03998759 and SAMN03998760 for SMTZ1-83, SMTZ1-45 and SMTZ-45 respectively.
Phylogenetic analyses
The concatenated ribosomal protein tree was generated using 16 syntenic genes that have been shown to undergo limited lateral gene transfer (rpL2, 3, 4, 5, 6, 14, 15, 16, 18, 22, 24 and rpS3, 8, 10, 17, 19; Sorek et al., 2007). The reference data sets were derived from the Phylosift database (Darling et al., 2014), with additional sets from the Joint Genomic Institute IMG database (Castelle et al., 2013). The presence of all 16 genes was not seen in the majority of sequences. The number of genes missing varied; however, the majority of loci present were consistent throughout the bins. Scaffolds containing <50% of the selected syntenic ribosomal protein genes were not included in the analyses. We searched NCBI to include reference amino-acid sequences for phylogenetic analyses. Amino-acid alignments of the individual genes were generated using MUSCLE (Edgar, 2004). Alignments were trimmed for poorly aligned regions and gaps using the BMGE tool (Criscuolo and Gribaldo, 2010; with the following settings: -m BLOSUM30 –g 0.5) and concatenated before inferences methods. The curated alignments were then concatenated for phylogenetic analyses. The ribosomal protein tree included 71 taxa and 2295 unambiguously aligned positions. The trees shown in all the figures were generated using maximum likelihood using RAxML (GTRGAMMA for 16S rRNA gene tree and PROTGAMMA for r-protein rate distribution models; Stamatakis, 2014). Bootstrap values were generated using IQ-Tree with the C60+LG mixture model for 10 000 ultra rapid bootstraps (Nguyen et al., 2015).
Results and Discussion
Genomic reconstruction of a novel, deeply branched archaeal lineage
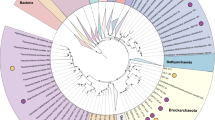
Metagenomic assembly and binning of 262 Gb of sequence data generated over 150 genomic bins from three sites, and distinct redox regimes, in the WOR estuary sediment (Baker et al., 2015). Large-scale microbial community analysis and single-cell genomic reconstruction was preformed on the data generated from these sites (Lazar et al., 2014; Baker et al., 2015). To begin to resolve the metabolic capabilities of uncultured and genomically unsampled phyla we generated a phylogenetic tree of 16S rRNA genes from all of the genomic bins (Figure 1). This analysis revealed a 3.31 Mb bin from site 1 (SMTZ1-83) belonging to a deeply branched, previously undescribed archaeal group (Raes et al., 2007). The genome is estimated to be 90.3% complete (Parks et al., 2015), with single copies of 137 of the 150 markers, multiple copies of eight other markers and minimal contamination (Table 1). Comparison between genomes showed that 58% of the genes are shared between all three bins and 84% are shared among two of them.

Phylogenetic position of the full-length 16S rRNA gene from the SMTZ1-83 genomic bin within the Archaea. The other two bins contain 16S rRNA gene <900 bp, thus were not included in this analysis. Bin SMTZ1-45 contains 682 bp of a 16S rRNA gene. The tree was generated using RAxML in the ARB software package (Ludwig et al., 2004). Closed and open circles represent bootstraps >80% and >60% (respectively) generated using IQtree (Nguyen et al., 2015). Closed circles represent bootstrap values of >70%. Giardia lamblia was used as the outgroup.
Due to the high conservation of 16S rRNA genes, these genes are often fragmented in short-read genomic libraries, and absent from bins (Miller et al., 2011). Therefore, in order to identify additional bins belonging to this group we generated concatenated ribosomal protein trees from all of the genomic bins. This revealed an additional bin, which contained fragments from two assemblies. These contigs from the two assemblies were separated into two bins, SMTZ1-45 with 3.87 Mb (from site 1) and SMTZ-45 with 2.37 Mb (from sites 2 and 3).
The 16S rRNA gene from bin SMTZ1-83 clustered with sequences recovered from a variety of environments including mangroves (Pires et al., 2012), freshwater Lake Pontchartrain (Amaral-Zettler et al., 2008) and hydrothermal marine sediments (Yanagawa et al., 2013a), suggesting that this group of organisms is broadly distributed in aquatic sediments. Samples collected at these sites revealed that sulfate reduction and methanogenic archaea dominate the sediments profiles similar to the WOR esturary. They have also been identified in hydrothermal vents in the Okinawa Trough (Pires et al., 2012; Moyer et al., 2013; Yanagawa et al., 2013b). To our knowledge, 16S rRNA sequences for these archaea were not identified from other estuaries. Rank abundance plots of the SMTZ communities revealed that these organisms are rare members of the WOR sediment community (Supplementary Figure S2).
Before this study this phylogenetic lineage had not even been given a candidate phylum designation (Yanagawa et al., 2013a), although both 16S rRNA gene phylogenies and the ribosomal protein trees revealed that these genomes are distinct from all previously described archaeal phyla. The lineage branched deeply within the TACK superphylum (Figure 1; Guy and Ettema, 2011) and shared a root with the newly described Lokiarchaeota (Spang et al., 2015) in the concatenated ribosomal protein gene tree (Figure 2). Furthermore, the novelty and deep branching is reflected in a fairly even distribution of taxonomic top hits to their genes between Archaea and Bacteria (Figure 3). Thus we propose that this group of archaea from the WOR sediments be given a distinct name that reflects its antiquity and sibling group relationship with the Lokiarchaeota, ‘Thorarchaeota’.

Phylogenetic analysis of 16 concatenated ribosomal proteins (rpL2, 3, 4, 5, 6, 14, 15, 16, 18, 22, 24 and rpS3, 8, 10, 17, 19) generated using RAxML in the ARB software package. Closed and open circles represent bootstraps >80% and >60% (respectively) generated using IQtree (Nguyen et al., 2015).

Domain-level taxonomic distribution of top hits to genes in the three Thorarchaeota genomes, based on nucleotide comparisons (BLASTn) of the genes with those present in Genbank (NCBI, nt database).
Carbon metabolism
Sediments receive a variety of forms of detrital organic matter from the overlying water column, which provide carbon and nitrogen to the benthic microbial communities. Consistent with recently obtained benthic archaeal genomes (Lloyd et al., 2013), Thorarchaeota have the genomic potential to degrade peptides (Figure 4; Supplementary Table S1). Genes for protein degradation and assimilation, including clostripain (cloSI) and gingipain (rgpA), were identified along with several other extracellular peptidases. Genes encoding a complete branched amino-acid importer (livKHMGF) were found in SMTZ1-83 and SMTZ1-45. Partial dipeptide (dppABCF) and oligopeptide (oppABCDF) importers were detected in all Thorarchaeota genomes, indicating that transport of peptides into the cell could be a common capability of these archaea. Numerous endopeptidase genes including pepT, pepA and pepB as well as multiple aminotransferase genes from families I through V (aspB, argD, glmS, puuE and serC) known to assist with the degradation of imported peptides to keto-acids were identified in all three bins of the Thorarchaeota. The abundance and variety of these peptidases in the Archaea suggest that proteins and peptides function as a likely carbon source.

Metabolic reconstruction of the Thorarchaeaota genomic bin (SMTZ1-83) based on genes identified using the KEGG database. Genes for various metabolisms, importers and hydrogenases are represented. White boxes represent the genes that are present in the genome and gray boxes represent genes that are absent. Details about the genes numbered are provided in Supplementary Table S2.
The Thorarchaeota genomic bins also encode putative oxidoreductases including pyruvate:ferredoxin oxidoreductase (porABDG), indolepyruvate ferredoxin oxidoreductase (iorAB) and aldehyde oxidoreductase (aor) that transform the keto-acids into acetyl-CoA and organic acids such as acetate (Lloyd et al., 2013). Multiple copies of all these genes were identified in SMTZ1-83, SMTZ-45 and SMTZ1-45 (Supplementary Tables S2 and S3). These enzymes require as electron acceptor oxidized ferredoxin, which can be supplied by Ni–Fe hydrogenases (Sapra et al., 2003). Genes coding for several Ni–Fe hydrogenases were found in the thorarchaeal genomes. A Phylip PROML phylogenetic tree of known Ni–Fe hydrogenases showed these genes grouped closely with several subunits of the methanogenesis-related F420-reducing and non-reducing dehydrogenases (Supplementary Figure S3). The lack of other genes required for methanogenesis suggests that these Ni–Fe hydrogenases are likely performing non-methanogenic functions in the Thorarchaeota.
Carbohydrates are another possible carbon source for heterotrophic microorganisms. Genomes were searched for pathways involved in carbohydrate degradation using the CAZy server (Lombard et al., 2014). A total of 81, 72 and 72 carbohydrate-active enzymes were found in SMTZ-45, SMTZ1-45 and SMTZ1-83 respectively. However only a small percentage (between 8.3% and 12.3%) of these enzymes could be assigned specific roles in carbohydrate degradation, mostly as cellulases and alpha-mannosidases (Supplementary Table S1). The limited quantity and constrained roles of carbohydrate degradation genes suggest that these archaea use carbohydrates selectively as carbon sources. The identification of an apparent complete peptide degradation pathway leads us to suggest that Thorarchaeota may prefer the heterotrophic degradation of proteins over carbohydrates.
The assembled genomes show the capability for glycolysis with all the required genes except hexokinase, present in at least one of the three genomes. Thorarchaeota appear to have the ability to convert glucose-6-phosphate partially or completely to phosphoenol pyruvate (Figure 4). Pyruvate kinase was not identified in any of the Thorarchaeota and instead pyruvate appears to be generated using phosphoenolpyruvate synthase (Bräsen et al., 2014). All three genomes contain the genes necessary to convert pyruvate to acetyl-CoA including both the E1 (PDHA) and E2 (PDHB) units of pyruvate dehydrogenase and all the subunits of pyruvate ferredoxin oxioreductase (porABDG). Although the majority of enzymes that are necessary for the citrate acid cycle were not present, a near-complete succinate hydrogenase complex was present in all three genomes. The genomic evidence suggests that terminal oxidation via the citric acid cycle is not a major energy source for the Thorarchaeota.
The genomes all possessed the ADP-forming subunit of acetyl-CoA synthetase that has been seen to catalyze acetate production in Pyrococcus furiosus and other archaea (Glasemacher et al., 1997). The presence of this enzyme suggests that acetyl-CoA could be converted to acetate; however genes for ethanol fermentation did not appear to be present. Therefore, we believe that Thorarchaeota are likely involved in the production of acetate through the hydrolytic cleavage of acetyl-CoA. As acetate is a key substrate for sulfate reduction and methanogenesis in estuary sediments (Oremland and Polcin, 1982) and appears to be an important link between fermentation and respiratory metabolisms (Wrighton et al., 2014), the ability of Thorarchaea to generate acetate could greatly influence the terminal anaerobic degradation cascade.
The Wood–Ljungdahl pathway enables autotrophic carbon fixation by converting extracellular two CO2 molecules first to acetyl-CoA and then to acetate. The first CO2 is converted to a methyl group through sequential transfer of six electrons. The methyl group is then transferred to convert a second CO2 to acetyl-CoA (Ragsdale and Pierce, 2008). Although SMTZ-45 did not show any genes for enzymes in this pathway, near-complete gene sets were found in the SMTZ1-45 and SMTZ1-83 bins. The CO2-reducing enzymes lacked formate dehydrogenase for the initial CO2 reduction step, and methylene-tetrahydrofolate reductase (metF), which converts 5,10-methylene-tetrahydrofolate to 5-methyl-tetrahydrofolate. Genes for acetyl-CoA synthesis were nearly complete except for the acsE gene, which codes for the methyltransferase that transfers the methyl group from methyl-tetrahydroformate to a corrinoid iron/sulfur protein, an intermediate methyl carrier; from here, the methyl group is accepted by the key enzyme of the pathway, CO-dehydrogenase/acetyl-CoA synthase, to produce acetyl-CoA and acetate (Ragsdale and Pierce, 2008). If the missing genes are accounted for by the incomplete sequence coverage of bins SMTZ1-45 and SMTZ1-83, these Thorarchaeaota may be able to use the Wood–Ljungdahl pathway of carbon fixation and support themselves by autotrophic acetogenesis, depending on the need. However, it is possible that the pathway is in fact incomplete in these archaea. The presence of this pathway is not direct evidence of autotrophy, as CO2 could serve primarily as an acceptor for electrons derived from a wide range of fermentative reactions within the organism, rather than an autotrophic carbon source that sustains the biosynthetic needs of the cell (Ragsdale and Pierce, 2008).
The possible role of Thorarchaeota in sulfur cycling
The microbial remineralization of organic matter in sediments is often coupled to anaerobic respiration of sulfate and nitrate/nitrite reduction in sediments (Jørgensen, 1982; Kostka et al., 2002). As archaea participate extensively in the cycling of nitrogen and sulfur (Canfield and Raiswell, 1999; Cabello et al., 2004), and previous 16S rRNA detection of the Thorarchaoeta in anaerobic aquatic sediments is broadly consistent with these geochemical roles, we searched the Thorachaeota genomic bins for the presence of relevant genes. All three of these archaeal bins have homologs to sulfohydrogenase, which has been shown to be involved in the reduction of elemental sulfur in the archaeon P. furiosus (Ma et al., 1993). We found that the Thorarchaeaota bins contain a variety of hydrogenase genes (9–15 per bin). In an attempt to resolve the functions of these proteins we compared them with other phylogenetically related proteins. All three genomic bins have a single copy of the alpha, beta, gamma and delta subunits (hydABGD) that are monophyletic with those sequences from P. furiosus (Supplementary Figure S3). Sequences that code for the reactive gamma subunit grouped closest to hypothetical proteins from the bacterial phyla Aminicenantes and Cloacimonetes (Rinke et al., 2013) and formed a monophyletic group with genes isolated P. furiosus. The beta sulfhydrogenases grouped with a beta subunit of the same gene from P. furiosus and a hydrogenase/sulfur reductase isolated from Pelodictyon phaeoclathratiforme, a member of the bacterial phylum Chlorobia (Supplementary Figure S3; Lucas et al., 2008). Various other Ni–Fe hydrogenases were also identified, however only the delta subunit of the F420-reducing hydrogenase is seen in all three genomes. As the Ni–Fe hydrogenases seen in these three archaeal genomes show close affiliations to the sulfhydrogenase subunits of P. furiosus, the Thorarchaeaota are most likely capable of sulfur reduction.
These genomes were isolated from the SMTZ where sulfate is almost completely depleted by sulfate reduction and sulfide concentrations are highest (Lazar et al., 2014). Of these genomic bins SMTZ-45 has the most well-defined sulfur metabolism with genes for a sulfate/thiosulfate importer, a thiosulfate reductase electron transporter (phsB) and the sulfhydrogenase. SMTZ1-83 only appears to have the sulfhydrogenase, whereas SMTZ1-45 has both the sulfhydrogenase and a thiosulfate reductase, suggesting that these archaea might also be mediating the transformation of intermediate sulfur compounds (Supplementary Table S3). For both SMTZ-45 and SMTZ1-45 the closest NCBI hits to the thiosulfate reductase included a 4Fe–4S ferredoxin isolated from Desulfobacterium autotrophicum (Brysch et al., 1987), an Enterobacteriacea thiosulfate reductase electron transporter (phsB) (Brenner, 1983) and the polysulfide reductase subunit B of Citrobacter freundii (Sakazaki, 1984). The phs operon codes for a molybdopterin cofactor that uses cysteine residues to bind and break the di-sulfur bonds (Hinsley and Berks, 2002). Homologs for these cysteine residues and for the iron sulfur clusters responsible for electron transfer have been identified on the phsB gene (Heinzinger et al., 1995). Although this gene has been seen in a few non-thiosulfate-reducing organisms, all BLAST hits to our sequences had functional enzymes (Park et al., 2012). Thiosulfate reductase preferentially utilizes thiosulfate as an electron acceptor. Thiosulfate is a dominant sulfur oxidation product in marine sediments (Jørgensen, 1990) and can be used as a substrate for both oxidation and reduction; microorganisms that reduce this important intermediate in the sulfur cycle would be imperative to sulfur transformation in anoxic environments (Stoffels et al., 2012). The presence of genes involved in both elemental sulfur and thiosulfate reduction suggests that these archaea may be key players in the reduction of intermediate sulfur species at the SMTZ redox layer.
Conclusions
Unlike many other methods for environmental community analysis, the ability to reconstruct unique genomes allows for insight into the ecological roles microbes have in the environment. Genomic reconstruction of Thorarchaeota, a widespread sediment group from the WOR estuary, has resolved the metabolic potential and previously unknown niches for these archaea, including their apparent ability to degrade organic matter, fix inorganic carbon, and reduce sulfur. They may be having an important role in the reduction of intermediate sulfur compounds generated by the oxidation of sulfide in the SMTZ (Lazar et al., 2014). Furthermore, these first genomic glimpses at the Thorarchaeota provide strong evidence that they are capable of acetogenesis. Based on energetic considerations it has been hypothesized that acetogens generally cannot outcompete methanogens, except in oligotrophic marine settings and the deep subsurface (Lever, 2012; Oren, 2012); targeted habitat studies should reveal whether Thorarchaeota match this prediction. Incorporation of this new knowledge into future models will result in a more accurate representation of benthic biogeochemical cycling. The acquisition of additional thorarchaeotal genomes, and matching gene expression studies, from other sediment environments will provide a better understanding of this previously overlooked archaeal phylum. Given the deep branching of the Thorarchaeota within the archaeal domain and its relationship to Lokiarchaeota, future evolutionary investigations of this phylum will provide insights into the ancestral relationships between the archaea and eukaryotes.
References
Amaral-Zettler LA, Rocca JD, Lamontagne MG, Dennett MR, Gast RJ . (2008). Changes in microbial community structure in the wake of Hurricanes Katrina and Rita. Environ Sci Technol 42: 9072–9078.
Baker BJ, Comolli LR, Dick GJ, Hauser LJ, Hyatt D, Dill BD et al. (2010). Enigmatic, ultrasmall, uncultivated Archaea. Proc Natl Acad Sci USA 107: 8806–8811.
Baker BJ, Dick GJ . (2013). Omic approaches in microbial ecology: charting the unknown. Microbe 8: 353–359.
Baker BJ, Lazar CS, Teske AP, Dick GJ . (2015). Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 3: 14.
Bauer JE, Cai W-J, Raymond PA, Bianchi TS, Hopkinson CS, Regnier PAG . (2013). The changing carbon cycle of the coastal ocean. Nature 504: 61–70.
Biddle JF, Lipp JS, Lever MA, Lloyd KG, Sørensen KB, Anderson R et al. (2006). Heterotrophic Archaea dominate sedimentary subsurface ecosystems off Peru. Proc Natl Acad Sci USA 103: 3846–3851.
Brasen C, Esser D, Rauch B, Siebers B . (2014). Carbohydrate metabolism in archaea: current insights into unusual enzymes and pathways and their regulation. Microbiol Mol Biol Rev 78: 89–175.
Brenner DJ . (1983). Opposition to the proposal to replace the family name Enterobacteriaceae. Int J Syst Bacteriol 33: 892–895.
Brysch K, Schneider C, Fuchs G, Widdel F . (1987). Lithoautotrophic growth of sulfate-reducing bacteria, and description of Desulfobacterium autotrophicum gen. nov., sp. nov. Arch Microbiol 148: 264–274.
Cabello P, Roldan MD, Moreno-Vivián C . (2004). Nitrate reduction and the nitrogen cycle in archaea. Microbiology 150: 3527–3546.
Cai W-J . (2011). Estuarine and coastal ocean carbon paradox: CO2 sinks or sites of terrestrial carbon incineration? Annu Rev Mar Sci 3: 123–145.
Canfield DE, Raiswell R . (1999). The evolution of the sulfur cycle. American J Sci 299: 697–723.
Castelle CJ, Hug LA, Wrighton KC, Thomas BC, Williams KH, Dongying W et al. (2013). Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat Commun 4: 2120.
Castelle CJ, Wrighton KC, Thomas BC, Hug LA, Brown CT, Wilkins MJ et al. (2015). Genomic expansion of domain Archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Current Biol 25: 690–701.
Criscuolo A, Gribaldo S . (2010). BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evolutionary Biol 10: 210.
Darling AE, Jospin G, Lowe E, Matsen FA, Bik HM, Eisen JA . (2014). Phylosift phylogenetic analysis of genomes and metagenomes. PeerJ 2: e243.
Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP et al. (2009). Community-wide analysis of genome sequence signatures. Gen Biol 10: R85.
Durbin AM, Teske A . (2011). Microbial diversity and stratification of South Pacific abyssal marine sediments. Environ Microbiol 12: 3219–3234.
Edgar R . (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1791.
Glasemacher J, Bock AK, Schmid R, Schönheit P . (1997). Purification and properties of acetyl-coa synthetase (adp-forming), an archaeal enzyme of acetate formation and ATP synthesis, from the hyperthermophile Pyrococcus furiosus. Eur J Biochem 244: 561–567.
Guy L, Ettema TJ . (2011). The archaeal ‘TACK’ superphylum and the origins of eukaryotes. Trends Microbiol 19: 580–587.
Heinzinger NK, Fujimoto SY, Clark MA, Moreno MS, Barrett EL . (1995). Sequence analysis of the phs operon in Salmonella typhimurium and the contribution of thiosulfate reduction to anaerobic energy metabolism. J Bacteriol 177: 2813–2820.
Hinsley AP, Berks BC . (2002). Specificity of respiratory pathways involved in the reduction of sulfur compounds by Salmonella enterica. Microbiology 148: 3631–3638.
Hug LA, Castelle CJ, Wrighton KC, Thomas BC, Sharon I, Frischkorn KR et al. (2013). Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1: 22.
Jørgensen BB . (1982). Mineralization of organic matter in the sea bed – the role of sulphate reduction. Nature 296: 643–645.
Jørgensen BB . (1990). A thiosulfate shunt in the sulfur cycle of marine sediments. Science 249: 152–154.
Kelley CA, Martens CS, Chanton JP . (1990). Variations in sedimentary carbon remineralization rates in the White Oak River Estuary, North Carolina. Limnol Oceanogr 35: 372–383.
Kelley CA, Martens CS, Ussler W III . (1995). Methane dynamics across a tidally flooded riverbank margin. Limnol Oceanogr 40: 1112–1129.
Kostka JE, Roychoudhury A, Van Cappellen P . (2002). Rates and controls of anaerobic respiration across spatial and temporal gradients in saltmarch sediments. Biogeochemistry 60: 49–76.
Kubo K, Lloyd KG, Biddle JF, Teske A, Amann R, Knittel K . (2012). Archaea of the Miscellaneous Crenarchaeotal Group are abundant, diverse and widespread in marine sediments. ISME J 6: 1949–1965.
Lazar CS, Biddle JF, Meador TB, Blair N, Hinrichs KU, Teske AP . (2014). Environmental controls on intragroup diversity of the uncultured benthic archaea of the miscellaneous Crenarchaeotal group lineage naturally enriched in anoxic sediments of the White Oak River estuary (North Carolina, USA). Environ Microbiol 17: 2228–2238.
Lever MA . (2012). Acetogenesis in the energy-starved deep biosphere – a paradox? Front Microbiol 2: 284.
Lloyd KG, Alperin MJ, Teske A . (2011). Environmental evidence for net methane production and oxidation in putative ANaerobic MEthanotrophic (ANME) archaea. Environ Microbiol 13: 2548–2564.
Lloyd KG, Schreiber L, Petersen DG, Kjeldsen KU, Lever MA, Steen AD et al. (2013). Predominant archaea in marine sediments degrade detrital proteins. Nature 496: 215–218.
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B . (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42: 490–495.
Lucas S, Copeland A, Lapidus A, Glavina del Rio T, Dalin E, Tice H et al. (2008). Complete sequence of Pelodictyon phaeoclathratiforme BU-1. EMBL/GenBank/DDBJ databases.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
Ma K, Schicho RN, Kelly RM, Adams MWW . (1993). Hydrogenase of the hyperthermophile Pyrococcus furiosus is an elemental sulfur reductase or sulfhydrogenase: evidence for a sulfur-reducing hydrogenase ancestor. Proc Natl Acad Sci USA 90: 5341–5344.
Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y et al. (2012). IMG, the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res 40: D115–D122.
Martens CS, Goldhaber MB . (1978). Early diagenesis in transitional sedimentary environments of the White Oak River Estuary, North Carolina. Limnol Oceanogr 23: 428–441.
Meng J, Xu J, Qin D, He Y, Xiao X, Wang F . (2014). Genetic and functional properties of uncultivated MCG archaea assessed by metagenome and gene expression analyses. ISME J 8: 650–659.
Miller CS, Baker BJ, Thomas BC, Singer S, Banfield JF . (2011). EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Gen Biol 12: R44.
Moyer CL, Birrien JL, Aoike K, Sunamura M, Urabe T, Mottl MJ et al. (2013). The first microbiological contamination assessment by deep-sea drilling and coring by the D/V Chikyu at the Iheya North hydrothermal field in the Mid-Okinawa Trough (IODP Expedition 331). Front Microbiol 4: 327.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ . (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32: 268–274.
Oremland RS, Polcin S . (1982). Methanogenesis and sulfate reduction: competitive and noncompetitive substrates in estuarine sediments. Appl Environ Microbiol 44: 1270–1276.
Oren A . (2012). There must be an acetogen somewhere. Front Microbiol 3: 22.
Park C-I, Choib SY, Kimb WS, Ohb MJ, Kimb EH, Kimb HY et al. (2012). Characterization of the unusual non-thiosulfate-reducing Edwardsiella tarda isolated from eel (Anguilla japonica farms. Aquaculture 356–357: 415–419.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW . (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Gen Res 25: 1043–1055.
Peng Y, Leung HCM, Yiu SM, Chin FYL . (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28: 1420–1428.
Pires AC, Cleary DF, Almeida A, Cunha A, Dealtry S, Mendonca-Hagler LC et al. (2012). Denaturing gradient gel electrophoresis and barcoded pyrosequencing reveal unprecedented archaeal diversity in mangrove sediment and rhizosphere samples. Appl Environ Microbiol 78: 5520–5528.
Raes J, Korbel JO, Lercher MJ, von Mering C, Bork P . (2007). Prediction of effective genome size in metagenomic samples. Genome Biol 8: R10.
Ragsdale SW, Pierce E . (2008). Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim Biophys Acta 1784: 1873–1898.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437.
Sakazaki R . (1984). Genus IV. Citrobacter Werkman and Gillen 1932, 173AL. In: Krieg NR, Holt JG (eds). Bergey's Manual of Systematic Bacteriology, vol. 2. The Williams & Wilkins Co: Baltimore, pp 458–461.
Sapra R, Bagramyan K, Adams M . (2003). A simple energy-conserving system: proton reduction coupled to proton translocation. Proc Natl Acad Sci USA 100: 7545–7550.
Seyler LM, McGuinness LM, Kerkhof LJ . (2014). Crenarchaeal heterotrophy in salt marsh sediments. ISME J 8: 1534–1543.
Sorek R, Zhu Y, Creevey CJ, Francino MP, Bork P, Rubin EM . (2007). Genome-wide experimental determination of barriers to horizontal gene transfer. Science 318: 1449–1452.
Spang A, Saw JH, Jorgensen SL, Zaremba-Niedzwiedzka K, Martijn J, Lind AE et al. (2015). Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521: 173–179.
Stamatakis A . (2014). RAxML Version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313.
Stoffels L, Krehenbrink M, Berks BC, Unden G . (2012). Thiosulfate reduction in Salmonella enterica Is driven by the proton motive force. J Bacteriol 194: 475–485.
Wrighton KC, Castelle CJ, Wilkins MJ, Hug LA, Sharon I, Thomas BC et al. (2014). Metabolic interdependencies between phylogenetically novel fermenters and respiratory organisms in an unconfined aquifer. ISME J 7: 1452–1463.
Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC et al. (2012). Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337: 1661–1665.
Yanagawa K, Morono Y, Beer DD, Haeckel M, Sunamura M, Futagami T et al. (2013a). Metabolically active microbial communities in marine sediment under high-CO2 and low-pH extremes. ISME J 7: 555–567.
Yanagawa K, Nunoura T, McAllister SM, Hirai M, Breuker A, Brandt L et al. (2013b). The first microbiological contamination assessment by deep-sea drilling and coring by the D/V Chikyu at the Iheya North hydrothermal field in the Mid-Okinawa Trough (IODP Expedition 331). Front Microbiol 4: 327.
Acknowledgements
The sequencing at JGI was funded by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 to BJB. Cassandre Lazar as well as sampling in the White Oak River estuary have been supported by the European Research Council ‘DARCLIFE’ grant 247153 to K-U Hinrichs. AT was in part supported by the Center for Dark Energy Biosphere Investigations (C-DEBI). We thank Dr Thijs JG Ettema for the naming of Thorarchaeota.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Seitz, K., Lazar, C., Hinrichs, KU. et al. Genomic reconstruction of a novel, deeply branched sediment archaeal phylum with pathways for acetogenesis and sulfur reduction. ISME J 10, 1696–1705 (2016). https://doi.org/10.1038/ismej.2015.233
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.233
This article is cited by
-
Inference and reconstruction of the heimdallarchaeial ancestry of eukaryotes
Nature (2023)
-
Unique mobile elements and scalable gene flow at the prokaryote–eukaryote boundary revealed by circularized Asgard archaea genomes
Nature Microbiology (2022)
-
Microbial diversity in extreme environments
Nature Reviews Microbiology (2022)
-
Three families of Asgard archaeal viruses identified in metagenome-assembled genomes
Nature Microbiology (2022)
-
A standardized archaeal taxonomy for the Genome Taxonomy Database
Nature Microbiology (2021)
