
Abstract
Cancer cachexia is a multifactorial syndrome that includes muscle wasting and inflammation. As gut microbes influence host immunity and metabolism, we investigated the role of the gut microbiota in the therapeutic management of cancer and associated cachexia. A community-wide analysis of the caecal microbiome in two mouse models of cancer cachexia (acute leukaemia or subcutaneous transplantation of colon cancer cells) identified common microbial signatures, including decreased Lactobacillus spp. and increased Enterobacteriaceae and Parabacteroides goldsteinii/ASF 519. Building on this information, we administered a synbiotic containing inulin-type fructans and live Lactobacillus reuteri 100-23 to leukaemic mice. This treatment restored the Lactobacillus population and reduced the Enterobacteriaceae levels. It also reduced hepatic cancer cell proliferation, muscle wasting and morbidity, and prolonged survival. Administration of the synbiotic was associated with restoration of the expression of antimicrobial proteins controlling intestinal barrier function and gut immunity markers, but did not impact the portal metabolomics imprinting of energy demand. In summary, this study provided evidence that the development of cancer outside the gut can impact intestinal homeostasis and the gut microbial ecosystem and that a synbiotic intervention, by targeting some alterations of the gut microbiota, confers benefits to the host, prolonging survival and reducing cancer proliferation and cachexia.
Similar content being viewed by others


Introduction
The trillions of microbes that reside in the gastrointestinal tract of mammals (that is, the gut microbiota) maintain a symbiotic relationship with their host. The gut microbiota has a critical role in biological processes such as maturation of the immune system and host metabolism (Hooper et al., 2012; Brestoff and Artis, 2013). Reciprocally, the host’s immune system affords protection against microbes and partially determines the composition of the gut microbiota. This is achieved in part through the creation of a barrier that involves several factors, including secretion of glycoproteins (mucins), expression of tight junction proteins, release of agents that prevent the translocation of gut microbes (for example, immunoglobulin A) and secretion of antimicrobial proteins such as regenerating islet-derived 3 gamma (Reg3γ), secretory group IIA phospholipase A2 (Pla2g2) and α-defensins (Bevins and Salzman, 2011; Hooper et al., 2012; Mukherjee and Hooper, 2015). The appreciation that the gut microbiota has an essential role in human and animal health has generated tremendous interest in strategies to modulate its composition and metabolic function to benefit the host. Prebiotics are one of these strategies. Prebiotics typically refer to non-digestible compounds that, by being substrates for microorganisms in the gut, modulate the composition and/or activity of the gut microbiota, thus conferring a beneficial physiological effect to the host (Gibson and Roberfroid, 1995; Gibson et al., 2004; Bindels et al., 2015). The modulation of the gut microbiome has shown benefits in several experimental disease models, for example, inflammatory bowel diseases, obesity, type 2 diabetes, hepatic steatosis, gastrointestinal cancers, and allergic airway disease (Schwabe and Jobin, 2013; Tilg and Moschen, 2014; Bindels et al., 2015).
The potential role of gut microbes in carcinogenesis and cancer progression in the gastrointestinal tract has been an object of research for decades (Schwabe and Jobin, 2013; Louis et al., 2014). Recently, it has been demonstrated that the microbiome could also impact the occurrence and progression of cancer at extra-intestinal sites (Schwabe and Jobin, 2013). Reciprocally, it has been shown in rodent models that the gut microbiota is altered by cancers that occur outside the gastrointestinal tract (Lin et al., 2012; Bindels et al., 2012a, b). Because the gut microbiota participates in the regulation of host immunity and metabolism, it is of crucial importance from a therapeutic perspective to determine whether it has a role in tumour progression and associated metabolic disorders (often referred to as cancer cachexia) and to further elucidate the mechanisms underlying such interactions.
Cachexia is a serious but often neglected consequence of many chronic diseases. Its prevalence ranges from 5% to 15% of patients in end-stage chronic heart failure up to 50–80% of patients with advanced cancer (Argiles et al., 2014; von Haehling and Anker, 2014). Cancer cachexia is characterized by a loss of fat and muscle masses that is often accompanied by anorexia and inflammation (Argiles et al., 2014). Cancer cachexia remains an unmet medical need, and its treatment will likely require a multimodal therapeutic approach that combines nutritional support with exercises and pharmacological agents targeting the breakdown of skeletal muscle and inflammation (Fearon et al., 2012, 2013). We have previously shown that the administration of specific Lactobacillus strains or non-digestible carbohydrates fermented by gut microbes can reduce systemic inflammation and/or cancer progression (Bindels et al., 2012a, b), suggesting that the gut microbiome might be a potential therapeutic target for cancer cachexia. These studies raised several questions: how is the composition of the gut microbiota affected in cancer cachexia? Do the gut microbiota and the intestine have a role in cancer cachexia? What are the mechanisms involved in this crosstalk?
In this study, we used next-generation sequencing, metabolomics and molecular profiling of the host to gain in-depth insight regarding the impact of cancer and cachexia on the composition of the gut microbiota and to identify pathways that are potentially involved in these gut microbiota–host interactions. Using these approaches, we identified bacterial taxa that were associated with alterations of the intestinal homeostasis. The information obtained in the first studies was used to design a dietary approach combining the oral administration of a bacterium that is known to be decreased in cancer cachexia (Lactobacillus reuteri) and a prebiotic (inulin-type fructans, ITF). We then determined the effect of this synbiotic treatment on lifespan and other physiological hallmarks of cancer cachexia.
Materials and methods
Cell culture
Colon carcinoma 26 (C26) cells were maintained in DMEM high-glucose medium supplemented with 10% fetal bovine serum (PAA clone, PAA, Pasching, Austria), 100 μg ml−1 streptomycin and 100 IU ml−1 penicillin (Gibco, Ghent, Belgium) at 37 °C with 5% CO2. The Pro-B lymphocyte (BaF3/BaF) cell line transfected with Bcr-Abl was maintained as previously described (Bindels et al., 2012a).
Bacterial culture and in vitro testing
L. reuteri was grown at 37 °C in MRS medium (Difco, Franklin Lakes, NJ, USA) until the stationary phase and then washed. For peripheral blood mononuclear cell (PBMC) stimulation, the bacteria were suspended in phosphate-buffered saline containing 20% glycerol and stored at −80 °C until use. For the in vivo experiments, bacterial culture was centrifuged, supernatant removed and pelleted bacteria stored at −80 °C. Bacterial numerations were checked after thawing, and the bacteria were suspended at an adequate concentration. PBMCs were isolated from the blood of healthy donors as described previously (Foligne et al., 2007). Briefly, after Ficoll gradient centrifugation (Pharmacia, Stockholm, Sweden), mononuclear cells were collected, washed in RPMI-1640 medium (Life Technologies, Ghent, Belgium), and adjusted to 2 × 106 cells per ml in RPMI supplemented with gentamicin (150 g ml−1), l-glutamine (2 mm), and 10% fetal calf serum (Gibco). PBMCs were stimulated with phosphate-buffered saline or bacteria (bacterium:cell ratio of 10:1) for 24 h at 37 °C with 5% CO2. The supernatants were collected and stored at −20 °C until cytokine measurements were performed using R&D Duoset ELISA kits (R&D, Minneapolis, MN, USA).
Animals
Mice from Charles River Laboratories (Chatillon-sur-Chalaronne, France) were housed at two mice per cage with a 12-h light/dark cycle and fed an irradiated normal chow diet (AO4-10, 2.9 kcal g−1, Safe, Augy, France). Six to ten mice were included in each group, as indicated in the legends.
C26 model
Either a saline solution or C26 cells (1 × 106 cells in 0.1 ml saline) were injected subcutaneously (intrascapular) in male CD2F1 mice (8 weeks old).
BaF model
Either a saline solution or BaF cells (1 × 106 cells in 0.1 ml saline) were injected into the tail vein of anesthetized female Balb/c mice (6 weeks old). The dietary restriction experiment is described in the Supplementary information. For the synbiotic treatment, 1 day after the BaF injection, the mice received Lactobacillus reuteri 100-23 (2 × 108 CFU per ml) with 0.2 g per day of ITF (oligofructose, Orafti p95, 1.5 kcal g−1, Beneo-Orafti, Oreye, Belgium) in the drinking water, as previously described (Bindels et al., 2012a, b). The drinking solutions were replaced every day, and food and water intake were recorded. Synbiotic mice consumed on average 5.8 × 108 CFU per day for the main experiment and 6.34 × 108 CFU per day for the survival experiment (Supplementary Figure S1). Energy intake was calculated taking into account the energy content of the diet and energy from ITF.
At the end of the experiment (11 days for the C26 model and 13 days for the BaF model), portal blood and tissue samples were harvested following anaesthesia (ketamine and xylazine, or isoflurane gas, Abbot, Wavre, Belgium). Portal blood was collected in EDTA tubes and centrifuged (13 000 g, 3 min). Tissues were weighed and frozen in liquid nitrogen. All of the samples were stored at −80 °C.
For the survival experiment, a separate set of mice was used. Half the mice received the synbiotic treatment, as described above. A morbidity score was performed on day 14 as described in the Supplementary information. Mice were monitored every 3–6 h from day 15 to day 19. All mice died from cancer, and therefore, no tissues and blood were withdrawn.
The experiments were approved by the ethics committee of the Université catholique de Louvain, and the housing conditions were consistent with the specifications of the Belgian Law of 29 May 3013 for the protection of laboratory animals (Agreement LA 1230314).
Tissue mRNA analyses
The isolation of RNA, preparation of complementary cDNA and real-time PCR were performed as previously described (Bindels et al., 2012a). The RNA quality was checked using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The primer sequences are detailed in Supplementary Table S1. Full description of the quantitative real-time PCR (qPCR) procedures is provided in the Supplementary information.
Gut microbiota analyses
Genomic DNA was extracted from the caecal content using a QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany), including a bead-beating step. Absolute quantification of the bacterial taxa was performed by qPCR (primers presented in Supplementary Table S1). Full description of the qPCR procedures is provided in the Supplementary information. Illumina sequencing was performed as previously described (Krumbeck et al., 2015). The V5-V6 region of the 16S rRNA gene was amplified by PCR with modified primers. The amplicons were purified, quantified and sequenced using an Illumina Miseq to produce 2x300-bp sequencing products at the University of Minnesota Genomics Center. Initial quality-filtering of the reads was conducted with the Illumina Software, yielding an average of 148 370 pass-filter reads per sample. Quality scores were visualized and reads were trimmed to 240 bp. The reads were merged with the merge-Illumina-pairs application (Eren et al., 2013). For samples with >50 000 merged reads, a subset of 50 000 reads was randomly selected using Mothur 1.32.1 (Schloss et al., 2009) to avoid large disparities in the number of sequences. Subsequently, the UPARSE pipeline implemented in USEARCH v7.0.1001 (Edgar, 2013) was used to further process the sequences. Putative chimaeras were identified against the Gold reference database and removed. Clustering was performed with a 98% similarity cutoff to designate operational taxonomic units (OTUs). Non-chimaeric sequences were also subjected to taxonomic classification using the RDP MultiClassifier 1.1 from the Ribosomal Database Project (Cole et al., 2014) for phylum to genus characterization of the faecal microbiome.
The phylotypes were computed as percent proportions based on the total number of sequences in each sample. Alpha- and beta-diversity indexes were calculated using QIIME (Caporaso et al., 2010). The LDA effect size was computed and plotted using LEfSe (Segata et al., 2011). The complete protocol, statistical analysis and accession numbers are provided in the Supplementary materials and methods.
Metabolic profiling by 1H NMR spectroscopy
In all, 50 μl of portal plasma was mixed with 10 μl of D2O, vortexed and centrifuged, and the supernatant was transferred into NMR capillary tubes. The complete protocol and statistical analysis are described in the Supplementary materials and methods.
Statistical analyses
The statistical significance of differences between groups was assessed using Student’s t-test when comparing two groups, or one-way ANOVA followed by Tukey’s multiple comparison post hoc test when comparing several groups. The data are presented as the mean±s.e.m. or as whiskers plots with minima and maxima. P<0.05 was considered as statistically significant. More information is provided in the Supplementary materials and methods.
Results
Cancer development alters the caecal microbiota
Two models of cancer cachexia were used: the BaF model and the C26 model. The BaF model involves the intravenous injection of Bcr-Abl-expressing lymphocytes, thereby mimicking acute leukaemia (Bindels et al., 2012b). The C26 model consists of a subcutaneous injection of colon 26 (C26) tumour cells (Sciorati et al., 2009; Talbert et al., 2014). Both models are characterized by a loss of fat mass and muscle atrophy, as published previously for the BaF model (Bindels et al., 2012a) and as shown in Supplementary Figure S2 for the C26 model. The caecal microbiota analysis by Illumina sequencing of 16S rRNA tags revealed profound shifts in the composition of the microbiota in both models, as evidenced by principal coordinates analysis of the Morisita-Horn beta-diversity at the OTU level (Figures 1a and b). The three first principal coordinates explained 74% and 95% of the variation for the BaF and C26 models, respectively. Alpha-diversity indexes of richness (observed species), or richness and evenness (Shannon), were decreased in BaF and C26 mice compared with their healthy counterparts (Figures 1c and d). In the BaF and C26 models, 25 and 54 taxa were affected, respectively. The changes common to both models encompassed a decrease in the Clostridiales order, Clostridia class, Lactobacillaceae family and Lactobacillus genus, and an increase in the Enterobacteriaceae family, Enterobacteriales order, Parabacteroides genus and Bacteroidetes phylum (Figures 1e and f). In the C26 model, using an OTU-based analysis, we identified 70 significantly affected phylotypes. Among others, OTU 17 and OTU 97 (both identified as Escherichia) were increased by 2698-fold and 4676-fold, respectively, while the relative abundance of OTU 2 (identified as Parabacteroides goldsteinii/ASF 519) increased from 1% to 30% of the total 16S rDNA sequences. OTU 54 (identified as Lactobacillus johnsonii/gasseri) showed a four-fold decrease (Supplementary Table S2). qPCR analysis of Enterobacteriaceae, P. goldsteinii/ASF 519 and Lactobacillus spp. shows trends similar to the pyrosequencing results (Supplementary Figure S3).

Shifts in the composition of the gut microbiota in the BaF and C26 models. (a, b) Principal coordinate analysis of the Morisita-Horn beta-diversity index computed based on the OTU table. (c, d) Alpha-diversity indexes in cancer-bearing mice vs controls (CT). The observed species represent an index of richness, and the Shannon index takes into account the richness and evenness. (e, f) LDA scores in red for the taxa enriched in cancer-bearing mice and in green for the taxa enriched in control mice. N=6–8, *P<0.05, **P<0.01, ***P<0.001.
Cachectic mice exhibited reduced food intake (Supplementary Figure S4). To explore whether the modification of the specific taxa was due to changes in food intake, healthy mice were dietary restricted to mimic the decreased food intake of the BaF mice, and the same taxa were analysed by qPCR (Supplementary Figure S4). This experiment allowed us to conclude that the changes in Lactobacillus and Enterobacteriaceae that occur in cancer and the associated cachexia were not due to decreased food intake.
This set of experiment led us to the conclusion that, in two mouse models of cancer and cachexia, the gut microbiota is impacted by the development of cancer outside the gastrointestinal tract.
A synbiotic approach reduces the progression of cancer and associated cachexia
To further investigate the therapeutic potential of the gut microbiota, we applied a synbiotic approach by combining ITF, a well-known prebiotic, and a Lactobacillus species. The rationale for the use of dietary lactobacilli was that these bacteria were consistently reduced during cachexia, especially in the BaF model. In addition, we demonstrated in a previous study that the administration of a mixture of L. gasseri 311476 and L. reuteri 100-23 reduced systemic inflammation and muscle atrophy in the BaF model (Bindels et al., 2012a). In vitro co-incubation of each bacterial strain with human PBMCs suggested that L. gasseri evoked pro-Th1 responses, whereas L. reuteri appeared to possess more anti-inflammatory properties (Figure 2). Therefore, L. reuteri 100-23 was selected for the synbiotic approach and combined with ITF.

L. gasseri evokes pro-Th1 responses, whereas L. reuteri appears to possess more anti-inflammatory properties. Cytokine levels in the medium after co-culture of human PBMC and Lactobacillus species (two independent experiments involving a total of nine PBMC donors). *P<0.01 vs medium, #P<0.01 vs L. gasseri 311476.
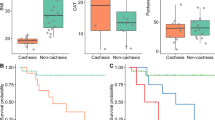
BaF mice were fed ITF and L. reuteri (LrI) throughout the course of the study. Thirteen days after injection, the LrI-treated mice exhibited decreased hepatic accumulation of BaF cells, as evidenced by a reduced liver weight and a lower expression of Bcr-Abl, a chimaeric protein that is specifically expressed in BaF cells (Figures 3a and b). Of note, we previously showed that these two parameters are key markers of BaF cell accumulation in the liver (Bindels et al., 2012b). Bcr-Abl expression in the spleen was not impacted by the synbiotic treatment (not detected in control mice, BaF: 1.00±0.14; BaF-LrI: 0.90±0.11, P-value=0.59, n=8). In addition, LrI resulted in the improvement of some cachectic features such as muscle mass loss and the expression of atrophy markers in tibialis and gastrocnemius muscles (Figures 3c and e). In this experiment, synbiotic treatment did not significantly impact the daily energy intake (Figure 3f).

The synbiotic approach reduces hepatic cancer cell accumulation and cachectic features in leukaemic mice. (a) Liver weight. (b) Bcr-Abl mRNA expression in the liver. ND, not detected. (c) Muscle weights. (d, e) mRNA expression of atrophy markers in the tibialis and gastrocnemius muscle. (f) Daily energy intake. (g) Morbidity score performed at day 14. (h) Kaplan–Meier curve showing the survival fractions analysed by the log-rank test (P=0.007). N=8 for (a–f) and n=10 for (g) and (h), *P<0.05 vs control, #P<0.05 vs BaF, ##P<0.01 vs BaF, $P=0.06 vs BaF.
A separate set of mice was used to investigate whether synbiotic treatment can affect morbidity and mortality. LrI administration lowered the morbidity score by half and prolonged the survival of mice by 2 days (Figures 3g and h). In this experiment, no change in energy intake was observed following the administration of LrI, except on days 1 and 12 (Supplementary Figure S5).
The synbiotic approach changes the gut microbiota composition
Synbiotic administration led to higher weights of caecal content and tissue (Supplementary Figure S5). Illumina sequencing confirmed profound shifts in the structure of the microbiota associated with cancer development, and it revealed that the synbiotic approach counteracted some of those changes (Figures 4 and 5, Supplementary Figure S5, Supplementary Tables S3 and S4). At the genus level, BaF injection was associated with the enrichment of Parabacteroides, Clostridium XI, Anaerotruncus and Escherichia, and the reduction of Barnesiella, Lactobacillus, Blautia, Clostridium XIVb and Parasutterella, among others; several of these changes were observed in previous experiments (Figures 4 and 5, Supplementary Figure S6 and Supplementary Table S3). LrI reversed the increased abundances of Escherichia and Anaerotruncus, whereas it increased the relative abundances of the Coriobacteriaceae family, Clostridium cluster XVIII, and some genera such as Barnesiella, Lactobacillus, Bifidobacterium and Parasutterella (Figures 4 and 5, Supplementary Figure S6). The OTU-based analyses confirmed these results and indicated a five-fold increase in the relative abundance of the segmented filamentous bacteria (SFB, OTU 185) (Figure 5, Supplementary Figure S7 and Supplementary Table S4). Quantification of Enterobacteriaceae, P. goldsteinii/ASF 519 and Lactobacillus spp. by qPCR confirmed the pyrosequencing results (Figures 5d–g).

The synbiotic approach changes the gut microbiota composition. (a) LEfSe cladogram in red for the taxa enriched in BaF mice and in green for the taxa enriched in control mice. (b) LEfSe cladogram in red for the taxa enriched in BaF mice and in green for the taxa enriched in BaF-LrI mice. N=8.

The synbiotic approach changes the abundance of several bacterial taxa. (a) Species enriched in BaF mice according to the LEfSe analysis. (b) Clostridium cluster XVIII. (c) SFB/OTU 185. (d–g) Bacteria levels in the caecal content of BaF mice fed or not the synbiotic, as determined by qPCR. AU, arbitrary units. N=8, *P<0.05 vs control, #P<0.05 vs BaF.
Of note, five of the eight bacterial taxa previously found to be commonly affected in both BaF and C26 models were similarly influenced by cancer development in this new set of experiments (namely, the Lactobacillaceae family, Lactobacillus genus, Enterobacteriaceae family, Enterobacteriales order and Parabacteroides genus) (Figure 4 and Supplementary Figure S6). This microbial signature (increase in the levels of Enterobacteriaceae and P. goldsteinii/ASF 519 and decrease in Lactobacillus levels) was also observed in two additional BaF experiments performed 18 months apart, demonstrating the reproducibility of our findings (Supplementary Figure S8).
Cancer progression changes the portal metabolome
To better understand how changing the composition and activity of the gut microbiota could reduce cancer cell proliferation and the underlying cachexia, we performed a metabolomics analysis of the portal blood of mice. Principal coordinate analysis of the metabolic profiles revealed clear clustering of the control mice due to the enrichment of glucose, fumarate and lipoproteins in the control group and of lactate, creatine and succinate in the BaF and BaF-LrI groups (Figure 6). A pairwise comparison of the NMR profiles of BaF and BaF-LrI mice did not highlight discriminant metabolites. O-PLS regressions on significantly affected OTUs and PCR transcripts revealed that plasma lipoproteins were negatively associated with the presence of some microorganisms, such as an unclassified Porphyromonadacae (OTU 88) and the Enterobacteriaceae family (Supplementary Figure S9).

Cancer progression changes the portal metabolome. (a) Principal coordinate analysis plot of the metabolomics profiles derived from portal plasma of control (blue), BaF (red) and BaF-LrI (green) mice, and associated loadings of PC2 (b) and PC1 (c). Pairwise O-PLS-DA comparison showing the calculated scores (x axis) against the cross-validated scores (y axis) and their associated loading coefficients. The weights of the variables are color-coded according to the square of the O-PLS-DA correlation coefficients. (d, e) Controls vs BaF and (f, g) controls vs BaF-LrI. N=6–8.
Synbiotic-mediated improvements in intestinal immune and barrier biomarkers correlate with microbial changes
To identify putative mechanistic links between microbial dysbiosis and cancer and cachexia, we characterized the gut microbiota–host crosstalk at the intestinal level. Antimicrobial peptides greatly affect the composition of the gut microbiota (Bevins and Salzman, 2011). Here, the expression of antimicrobial proteins such as Lysozyme 1, Reg3γ and Pla2g2 was decreased in BaF mice, as well as the expression of TCF4 (T cell-specific transcription factor 4), which drives Paneth cell differentiation (Figure 7a) (Bevins and Salzman, 2011). Whereas antimicrobials constitute a chemical barrier against pathogens and commensals, tight junction proteins, such as zonula occludens 1 (ZO-1) and occludin, and mucus secretion participate to construct a physical barrier that limits host exposure to gut microbes. BaF cell injection decreased the expression of occludin, zonula occludens 1 and Muc2 (Figure 7b). The expression levels of immune markers of inflammation (TNF-α, IFN-γ and IL-1β), the innate immune system (CD11b), proinflammatory macrophages (CD11c), T lymphocytes (CD3γ), Th1 lymphocytes (Tbet) and Th17 lymphocytes (IL-17A) were also decreased in the presence of cancer, whereas IL-6 and the macrophage pan-markers F4/80 and CD68 remained unaffected (Figures 7c and d). Interestingly, the administration of LrI counteracted most of the BaF-induced alterations detected in the intestine. Although ITF have been shown to improve gut barrier function through an increase in the production of glucagon-like peptide 2 (Cani et al., 2009), LrI administration did not increase the mRNA expression of proglucagon, the precursor of glucagon-like peptide 2 in this study (Figure 7b).

The synbiotic approach improves intestinal immune and barrier biomarkers in leukaemic mice. (a) mRNA expression of markers involved in Paneth cell differentiation and antimicrobial proteins. (b) mRNA expression of markers involved in gut permeability. (c) mRNA expression of immune markers related to inflammation, innate immunity (CD11b) and macrophages. (d) mRNA expression of immune markers related to lymphocytes (CD3γ) Th1 (Tbet), Th17 (IL-17A) and Treg (Foxp3, IL-10, Ebi3) cells. N=7–8, *P<0.05 vs control, #P<0.05 vs BaF.
Regulatory T (Treg) cells, a subset of lymphocytes that restrain the immune response, contribute to intestinal and systemic immune homeostasis and can be induced by commensal microbiota (Rubtsov et al., 2008; Arpaia et al., 2013; Atarashi et al., 2013). Treg development is controlled by the transcription factor Foxp3, while the function of Tregs is mediated by IL-35 and IL-10 (Collison et al., 2007; Gavin et al., 2007). Intestinal Foxp3 and IL-10 expression was reduced in BaF mice and restored following LrI treatment, whereas the expression of Ebi3, a subunit of the heterodimeric cytokine IL-35, was not impacted by cancer but was induced in response to LrI (Figure 7d). The immunomodulatory effect of LrI was not limited to the gastrointestinal tract. Indeed, LrI administration increased Foxp3 mRNA levels in the spleen (BaF: 1.0±0.1; BaF-LrI: 2.0±0.4, P=0.047), and this increase correlated with the relative abundance of Clostridium Cluster XVIII (Pearson r=0.71, P=0.002), a group of microbes that is able to induce Tregs (Atarashi et al., 2013).
Finally, a multiple correlation analysis between intestinal biomarkers and phylotypes that were significantly affected highlighted Escherichia sp. and P. goldsteinii/ASF 519 as two bacteria that correlated inversely with intestinal alterations. In addition, several OTUs, encompassing mainly Barnesiella, Lactobacillus, Enterorhabdus and unclassified Porphyromonadaceae species, correlated with Reg3γ, CD11b, CD11c and CD3γ expression (Figure 8, Supplementary Table S5).

Synbiotic-mediated improvements in intestinal immune and barrier biomarkers correlate with microbial changes. Heat map representation of the Pearson’s r correlation coefficient between intestinal biomarkers and significantly affected phylotypes. The closest known taxon is indicated with the OTU number within brackets. *Adjusted P<0.05.
Discussion
In this study, we demonstrated that the gut microbial balance was altered by the presence of tumours located outside the gastrointestinal tract, consistently with our previous study where we provided the first evidence of a modified gut microbiome in a mouse model of leukaemia and cachexia (BaF) (Bindels et al., 2012a). Caecal Lactobacillus spp. levels were decreased in leukaemic mice with cachexia, and a PCR-DGGE (denaturing gradient gel electrophoresis) analysis suggested that bacteria other than lactobacilli were affected by the disease. The same year, Lin et al. (2012) reported that subcutaneous implantation of colon cancer cells in rats was associated with a one-log increase in Enterobacteriaceae and Clostridium cluster I and XI in the faeces. In both studies, microbial dysbiosis was assessed using molecular approaches with limitation in dynamic range. Here we used a community-wide molecular approach to deeply characterize the microbial shifts occurring in two mouse models of cancer cachexia. Our data revealed a microbial signature common to both models that included the Enterobacteriaceae family/Escherichia genus, Lactobacillus genus and Parabacteroides goldsteinii/ASF 519 species. Some of these changes have also been observed in the previous studies (Lin et al., 2012; Bindels et al., 2012a), and have been reported as a sign of reduced colonization resistance (Lawley and Walker, 2013).
One potential mechanism for the dysbiosis observed during cachexia in mice might be the associated reduction in food intake observed at the late stage of the disease. Food intake has been reported to influence the composition of the gut microbiota (Crawford et al., 2009), and it may be considered that this reduced food intake constitutes a confounding factor in this mouse model. However, one must consider that anorexia is a fully fledged hallmark of cachexia, and thus, it should be integrated into experimental models. Our experiments revealed that the major shifts (Enterobacteriaceae and lactobacilli) occurred independently of food intake, suggesting that the presence of cancer cells impacts the gut microbial ecology independently of food intake. Nonetheless, even if anorexia contributes to dysbiosis, it would not impair the therapeutic implication of our findings. Synbiotic treatment modified to a very limited extent daily energy intake of cachectic mice in one out of two experiments. We can consider that the impact is minor compared with the leukaemia-induced reduced energy intake and likely not relevant enough to be taken into consideration for the interpretation of the data.
Our study results clearly demonstrated gut microbiota alterations in two different models of cachexia. These findings raise the question of whether this dysbiosis contributes to the observed pathologies. We have previously demonstrated the therapeutic interest of the restoration of lactobacilli (Bindels et al., 2012a). To our knowledge, the role of microorganisms, such as Escherichia and P. goldsteinii/ASF 519, in cancer cachexia has not been investigated yet. P. goldsteinii/ASF 519 is a mouse commensal species that is also found at low levels in humans (Dewhirst et al., 1999; Song et al., 2005). The ELDERMET study (Claesson et al., 2012) indicated that individuals in long-term care developed dominant levels of Parabacteroides and Alistipes that were associated with changes in frailty, inflammation and altered abundances of short-chain fatty acid producers. Similarly to P. goldsteinii, the genus Escherichia encompasses rodent and human commensal strains and is present at low levels in healthy subjects. Some strains are beneficial to the host, whereas others have been associated with inflammatory bowel disease and colorectal cancer (Manichanh et al., 2012; Louis et al., 2014; Sha et al., 2014). In our study, the synbiotic approach reduced the levels of Enterobacteriaceae. This phenomenon could be mediated by short-chain fatty acids (acetate, propionate and butyrate), the production of which is fostered by ITF (Bindels et al., 2012b; Everard et al., 2014), and which, in association with a decreased caecal pH, create a non-permissive environment for colitogenic Enterobacteriaceae (Veiga et al., 2010).
Additional research is needed to establish whether the gut microbiota has a role in cancer cachexia. Yet, based on our findings, the following hypothesis can be formulated. Systemic inflammation due to cancer development will alter the gut permeability, antimicrobial defence and Paneth cell function, leading to dysbiosis. Thereafter, the altered intestinal homeostasis, microbial dysbiosis and cancer cachexia would interact in a vicious cycle, where cancer-induced alterations of intestinal homeostasis foster the growth of certain bacteria that further increase systemic inflammation and cachexia. This hypothesis is supported by several elements, independently of the data presented herein. First, systemic inflammation, which has a key role in cancer cachexia (Fearon et al., 2012, 2013; Argiles et al., 2014; von Haehling and Anker, 2014), is present in BaF and C26 mice (Fearon et al., 2012; Bindels et al., 2012a, b) and increases epithelial permeability (Ivanov et al., 2010). Moreover, inflammation boosts the growth of Enterobacteriaceae and E. coli and creates an opportunity for E. coli to adhere to the colonic mucosa by decreasing the production of mucins and antimicrobials (Lupp et al., 2007; Winter et al., 2013; Louis et al., 2014). Finally, a similar relationship among Paneth cells, the loss of bacterial diversity, E. coli blooming and immune alterations has recently been reported in other mouse models (Inaba et al., 2010; Arthur et al., 2012; Eriguchi et al., 2012).
In addition to establishing the presence of microbial shifts in cancer cachexia, the results of the present study clearly provided evidence for considering the gut microbiota as a therapeutic target in cancer and cachexia. Indeed, nutritional modulation of the microbiome by combined pre and probiotic approaches resulted in the reduction of cancer cell proliferation and an improvement of the cachectic features, thus leading to prolonged survival. As L. reuteri is not able to metabolize ITF, this combination would be called a complementary synbiotic approach (Kolida and Gibson, 2011). Our data obtained in vitro confirmed that L. reuteri 100-23 is rather anti-inflammatory. Therefore, one could speculate that such immune modulation could foster cancer proliferation (Mantovani et al., 2008). However, this was not observed here.
To unravel the mechanisms by which the synbiotic approach provides its benefits, we performed 16S sequencing of the gut microbiota, metabolomic analysis of the portal blood and molecular characterization at the intestinal level, which allowed us to draw some hypotheses regarding the mechanism(s) underlying the benefits of the synbiotic treatment.
First, the synbiotic approach could mediate its benefits through the restoration of intestinal homeostasis. The implication of elevated gut permeability in cancer cachexia has been recently proposed (Puppa et al., 2011; Bindels and Delzenne, 2013). Our data clearly showed that cancer cells implanted at a distance from the gastrointestinal tract can affect the gut barrier and immune function. The changes in gut barrier function may contribute to the inflammatory status and thereby to the cachectic phenotype. By restoring the gut barrier function, the synbiotic treatment may impact systemic inflammation, as described in other pathological contexts (Cani et al., 2009).
Second, the synbiotic approach could modulate immunity by increasing the abundance of bacterial taxa with immunomodulatory properties, such as Clostridium cluster XVIII, the SFB and Lactobacillus reuteri 100-23. Atarashi et al. (2013) have previously demonstrated that a selected mixture of Clostridia strains falling within Clostridium clusters IV, XIVa and XVIII was able to induce Tregs. In accordance with this result, we found a strong correlation between Clostridium cluster XVIII and the expression of Foxp3 in the intestine and in the spleen. The SFB have a unique ability to educate the gut immune system and to induce a healthy state of physiological inflammation, notably through the IL-17 pathway (Ivanov et al., 2009; Schnupf et al., 2015). Accordingly, intestinal IL-17A expression increased in response to the synbiotic treatment. Finally, because L. reuteri 100-23 caused a slight increase in the numbers of Tregs in the spleen of Lactobacillus-free mice (Livingston et al., 2010), the synbiotic-induced increase in Foxp3 expression may also be directly mediated by L. reuteri 100-23.
The portal metabolome reflected mainly a state of energy demand in leukaemic mice, where significantly decreased glucose and lipoproteins levels, concomitant with increased levels of creatine and lactate, were observed. Increased creatine levels are typical of muscle wasting and consistent with the cachectic model. The increased lactate level in the portal plasma of leukaemic mice was likely a consequence of lactate production by BaF cells as previously reported (Bindels et al., 2012b). Succinate, an intermediate product in the Krebs cycle of endogenous and microbial origin, was also enriched in leukaemic mice. The metabolomic analysis did not highlight any portal metabolite discriminant for the synbiotic intervention. The lack of discriminant metabolites reinforces the hypothesis that the synbiotic approach provides a benefit via subtle changes in immunity or by impacting metabolic pathways that produce metabolites in low levels, such as propionate, which although detected, was below the limit of quantification in this study. Indeed, we have previously provided strong evidence to suggest that propionate produced through the bacterial fermentation of ITF reduces the proliferation of BaF cells in the liver (Bindels et al., 2012b).
The present study demonstrated that cancer cachexia was associated with profound alterations of the gut microbiota in two animal models. Our data further showed that dietary modulation of the gut microbiota that mitigated some of the dysbiotic patterns restored the intestinal gut barrier and immune function, thereby reducing cancer and associated cachexia. In addition, the synbiotic approach prolonged survival, a relevant primary outcome for nutrition therapy in cancer (Baracos et al., 2012). As all of the animal models have their limitations, exploration of the gut microbial ecosystem in cancer patients with cachexia is now needed before any extrapolation to humans. Which doses of synbiotics would have to be used in clinical trials to translate our findings remains highly speculative. Useful information can be obtained from published works, mainly in the context of metabolic alterations linked to obesity, which pointed out changes in the gut microbiota similar in mice and obese women upon prebiotic consumption that were associated with improvement of host metabolism (Dewulf et al., 2013).
Together, these data suggest a role for the gut microbiota and the intestinal barrier in the therapeutic management of cancer and associated cachexia. Future studies will be focused on investigating the causative role of specific gut microbes and altered intestinal homeostasis in the progression of cancer and associated cachexia, and assess the relevance of our findings in cancer patients with cachexia.
References
Argiles JM, Busquets S, Stemmler B, Lopez-Soriano FJ . (2014). Cancer cachexia: understanding the molecular basis. Nat Rev Cancer 14: 754–762.
Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P et al. (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504: 451–455.
Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ et al. (2012). Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338: 120–123.
Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500: 232–236.
Baracos VE, Pichard C, Attaix D . (2012). Survival: the relevant primary outcome for nutrition therapy in cancer patients. Curr Opin Clin Nutr Metab Care 15: 211–212.
Bevins CL, Salzman NH . (2011). Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol 9: 356–368.
Bindels LB, Beck R, Schakman O, Martin JC, De Backer FC, Sohet FM et al. (2012a). Restoring Specific lactobacilli levels decreases inflammation and muscle atrophy markers in an acute leukemia mouse model. PLoS One 7: e37971.
Bindels LB, Delzenne NM, Cani PD, Walter J . (2015). Towards a more comprehensive concept for prebiotics. Nat Rev Gastroenterol Hepatol 12: 303–310.
Bindels LB, Delzenne NM . (2013). Muscle wasting: The gut microbiota as a new therapeutic target? Int J Biochem Cell Biol 45: 2186–2190.
Bindels LB, Porporato P, Dewulf EM, Verrax J, Neyrinck AM, Martin JC et al. (2012b). Gut microbiota-derived propionate reduces cancer cell proliferation in the liver. Br J Cancer 107: 1337–1344.
Brestoff JR, Artis D . (2013). Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol 14: 676–684.
Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O et al. (2009). Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58: 1091–1103.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Claesson MJ, Jeffery IB, Conde S, Power SE, O'Connor EM, Cusack S et al. (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature 488: 178–184.
Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y et al. (2014). Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42: D633–D642.
Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM et al. (2007). The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 450: 566–569.
Crawford PA, Crowley JR, Sambandam N, Muegge BD, Costello EK, Hamady M et al. (2009). Regulation of myocardial ketone body metabolism by the gut microbiota during nutrient deprivation. Proc Natl Acad Sci USA 106: 11276–11281.
Dewhirst FE, Chien CC, Paster BJ, Ericson RL, Orcutt RP, Schauer DB et al. (1999). Phylogeny of the defined murine microbiota: altered Schaedler flora. Appl Environ Microbiol 65: 3287–3292.
Dewulf EM, Cani PD, Claus SP, Fuentes S, Puylaert PG, Neyrinck AM et al. (2013). Insight into the prebiotic concept: lessons from an exploratory, double blind intervention study with inulin-type fructans in obese women. Gut 62: 1112–1121.
Edgar RC . (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10: 996–998.
Eren AM, Vineis JH, Morrison HG, Sogin ML . (2013). A filtering method to generate high quality short reads using illumina paired-end technology. PLoS One 8: e66643.
Eriguchi Y, Takashima S, Oka H, Shimoji S, Nakamura K, Uryu H et al. (2012). Graft-versus-host disease disrupts intestinal microbial ecology by inhibiting Paneth cell production of alpha-defensins. Blood 120: 223–231.
Everard A, Lazarevic V, Gaia N, Johansson M, Stahlman M, Backhed F et al. (2014). Microbiome of prebiotic-treated mice reveals novel targets involved in host response during obesity. ISME J 8: 2116–2130.
Fearon K, Arends J, Baracos V . (2013). Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 10: 90–99.
Fearon KC, Glass DJ, Guttridge DC . (2012). Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 16: 153–166.
Foligne B, Nutten S, Grangette C, Dennin V, Goudercourt D, Poiret S et al. (2007). Correlation between in vitro and in vivo immunomodulatory properties of lactic acid bacteria. World J Gastroenterol 13: 236–243.
Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA et al. (2007). Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445: 771–775.
Gibson GR, Probert HM, Loo JV, Rastall RA, Roberfroid MB . (2004). Dietary modulation of the human colonic microbiota: updating the concept of prebiotics. Nutr Res Rev 17: 259–275.
Gibson GR, Roberfroid MB . (1995). Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J Nutr 125: 1401–1412.
Hooper LV, Littman DR, Macpherson AJ . (2012). Interactions between the microbiota and the immune system. Science 336: 1268–1273.
Inaba Y, Ashida T, Ito T, Ishikawa C, Tanabe H, Maemoto A et al. (2010). Expression of the antimicrobial peptide alpha-defensin/cryptdins in intestinal crypts decreases at the initial phase of intestinal inflammation in a model of inflammatory bowel disease, IL-10-deficient mice. Inflamm Bowel Dis 16: 1488–1495.
Ivanov AI, Parkos CA, Nusrat A . (2010). Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol 177: 512–524.
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485–498.
Kolida S, Gibson GR . (2011). Synbiotics in health and disease. Annu Rev Food Sci Technol 2: 373–393.
Krumbeck JA, Maldonado-Gomez MX, Martinez I, Frese SA, Burkey TE, Rasineni K et al. (2015). Introducing the concept of In Vivo Selection to identify bacterial stains with enhanced ecological performance when used in synbiotic applications. Appl Environ Microbiol 81: 2455–2465.
Lawley TD, Walker AW . (2013). Intestinal colonization resistance. Immunology 138: 1–11.
Lin XB, Dieleman LA, Ketabi A, Bibova I, Sawyer MB, Xue H et al. (2012). Irinotecan (CPT-11) Chemotherapy Alters Intestinal Microbiota in Tumour Bearing Rats. PLoS One 7: e39764.
Livingston M, Loach D, Wilson M, Tannock GW, Baird M . (2010). Gut commensal Lactobacillus reuteri 100-23 stimulates an immunoregulatory response. Immunol Cell Biol 88: 99–102.
Louis P, Hold GL, Flint HJ . (2014). The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12: 661–672.
Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC et al. (2007). Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2: 204.
Manichanh C, Borruel N, Casellas F, Guarner F . (2012). The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol 9: 599–608.
Mantovani A, Allavena P, Sica A, Balkwill F . (2008). Cancer-related inflammation. Nature 454: 436–444.
Mukherjee S, Hooper LV . (2015). Antimicrobial defense of the intestine. Immunity 42: 28–39.
Puppa MJ, White JP, Sato S, Cairns M, Baynes JW, Carson JA . (2011). Gut barrier dysfunction in the Apc(Min/+) mouse model of colon cancer cachexia. Biochim Biophys Acta 1812: 1601–1606.
Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X et al. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28: 546–558.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Schnupf P, Gaboriau-Routhiau V, Gros M, Friedman R, Moya-Nilges M, Nigro G et al. (2015). Growth and host interaction of mouse segmented filamentous bacteria in vitro. Nature 520: 99–103.
Schwabe RF, Jobin C . (2013). The microbiome and cancer. Nat Rev Cancer 13: 800–812.
Sciorati C, Touvier T, Buono R, Pessina P, Francois S, Perrotta C et al. (2009). Necdin is expressed in cachectic skeletal muscle to protect fibers from tumor-induced wasting. J Cell Sci 122: 1119–1125.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol 12: R60.
Sha S, Xu B, Kong X, Wei N, Liu J, Wu K . (2014). Preventive effects of Escherichia coli strain Nissle 1917 with different courses and different doses on intestinal inflammation in murine model of colitis. Inflamm Res 63: 873–883.
Song Y, Liu C, Lee J, Bolanos M, Vaisanen ML, Finegold SM . (2005). "Bacteroides goldsteinii sp. nov." isolated from clinical specimens of human intestinal origin. J Clin Microbiol 43: 4522–4527.
Talbert EE, Metzger GA, He WA, Guttridge DC . (2014). Modeling human cancer cachexia in colon 26 tumor-bearing adult mice. J Cachexia Sarcopenia Muscle 5: 321–328.
Tilg H, Moschen AR . (2014). Microbiota and diabetes: an evolving relationship. Gut 63: 1513–1521.
Veiga P, Gallini CA, Beal C, Michaud M, Delaney ML, DuBois A et al. (2010). Bifidobacterium animalis subsp. lactis fermented milk product reduces inflammation by altering a niche for colitogenic microbes. Proc Natl Acad Sci USA 107: 18132–18137.
von Haehling S, Anker SD . (2014). Prevalence, incidence and clinical impact of cachexia: facts and numbers-update 2014. J Cachexia Sarcopenia Muscle 5: 261–263.
Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM et al. (2013). Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339: 708–711.
Acknowledgements
We thank Véronique Peucelle, Remi Selleslagh and Bouazza Es Saadi for their skilled technical assistance and Alexandra Collignon, Mireille Alhouayek, Florence Sohet, Céline Druart, Emilie Catry, Nuria Salazar Garzo and Maud Alligier for help with the in vivo experiments and tissue sampling. The C26 cell line was a kind gift from Dr Mario Colombo, Fondazione IRCCS Istituto Nazionale Tumori, Italy. We thank the University of Minnesota Genomics Center for performing the pyrosequencing. LBB and PDC are, respectively, a Postdoctoral Researcher and a Research Associate from the F.R.S.-FNRS (Fond National de la Recherche Scientifique, Belgium). NMD and PDC are recipients of grants from FNRS. PDC is a recipient of the ERC Starting Grant 2013 (European Research Council, Starting grant 336452-ENIGMO).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Bindels, L., Neyrinck, A., Claus, S. et al. Synbiotic approach restores intestinal homeostasis and prolongs survival in leukaemic mice with cachexia. ISME J 10, 1456–1470 (2016). https://doi.org/10.1038/ismej.2015.209
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.209
This article is cited by
-
Cancer-associated cachexia — understanding the tumour macroenvironment and microenvironment to improve management
Nature Reviews Clinical Oncology (2023)
-
Gut microbiome and nutrition-related predictors of response to immunotherapy in cancer: making sense of the puzzle
BJC Reports (2023)
-
Gut OncoMicrobiome Signatures (GOMS) as next-generation biomarkers for cancer immunotherapy
Nature Reviews Clinical Oncology (2023)
-
Probiotics Attenuate Myopathic Changes in Aging Rats via Activation of the Myogenic Stellate Cells
Probiotics and Antimicrobial Proteins (2023)
-
The effects of synbiotic supplementation on enteral feeding tolerance, protein homeostasis, and muscle wasting of critically ill adult patients: a randomized controlled trial
Trials (2022)

{kind=link}