
Abstract
The CRISPR (clustered, regularly, interspaced, short, palindromic repeats)–Cas (CRISPR-associated genes) systems of archaea and bacteria provide adaptive immunity against viruses and other selfish elements and are believed to curtail horizontal gene transfer (HGT). Limiting acquisition of new genetic material could be one of the sources of the fitness cost of CRISPR–Cas maintenance and one of the causes of the patchy distribution of CRISPR–Cas among bacteria, and across environments. We sought to test the hypothesis that the activity of CRISPR–Cas in microbes is negatively correlated with the extent of recent HGT. Using three independent measures of HGT, we found no significant dependence between the length of CRISPR arrays, which reflects the activity of the immune system, and the estimated number of recent HGT events. In contrast, we observed a significant negative dependence between the estimated extent of HGT and growth temperature of microbes, which could be explained by the lower genetic diversity in hotter environments. We hypothesize that the relevant events in the evolution of resistance to mobile elements and proclivity for HGT, to which CRISPR–Cas systems seem to substantially contribute, occur on the population scale rather than on the timescale of species evolution.
Similar content being viewed by others


Introduction
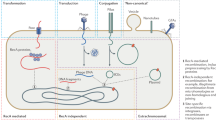
Archaea and bacteria encode diverse defense systems that protect these microbes from viruses, plasmids and other selfish genetic elements. One of the most efficient and widespread types of defense system is CRISPR (clustered, regularly, interspaced, short, palindromic repeats)–Cas (CRISPR-associated genes). CRISPR–Cas systems provide acquired, heritable immunity to bacteria and archaea against both viral (Barrangou et al., 2007; Manica et al., 2011) and plasmid (Marraffini and Sontheimer, 2008; Gudbergsdottir et al., 2011) invasion. CRISPR–Cas systems are more common in archaea than in bacteria, and are more abundant and contain more spacers in thermophiles than in mesophiles (Anderson et al., 2011; Weinberger et al., 2012b).
The CRISPR–Cas loci encompass arrays of direct, often partially palindromic repeats that are interspersed by short unique DNA segments (20–50 bp long), termed spacers, and multiple cas genes that encode proteins involved in the immune response. A seminal discovery that led to the characterization of the unique mechanisms of CRISPR–Cas action has been that some of the spacers are (nearly) identical to sequences of selfish elements (in this case, known as protospacers). Subsequently, it has been shown that a complex of Cas1 and Cas2 proteins excises protospacer DNA from invading elements and integrates them into the CRISPR arrays (Barrangou et al., 2007; Deveau et al., 2008; Li et al., 2014). One or several spacer arrays in a prokaryotic cell can be transcribed and processed into small CRISPR RNA (crRNA) molecules by a complex of several Cas proteins known as Cascade. The Cascade complex then mediates the formation of a duplex between the crRNA and the cognate protospacer sequence in an invading nucleic acid molecule triggering degradation of the latter (Haurwitz et al., 2010; Semenova et al., 2011).
The fact that the CRISPR–Cas systems are able to continuously acquire new spacers enables partial reconstruction of the history of past selfish-element infections (Tyson and Banfield, 2008; Denef et al., 2010; Held et al., 2010; Stern et al., 2012; Weinberger et al., 2012a). In the absence of parasitic elements, spacers can be easily lost owing to the deletion bias of prokaryotic genome evolution (Kuo and Ochman, 2009) and the presumed cost of maintaining CRISPR systems (Weinberger et al., 2012a).
The source(s) of this putative cost of the CRISPR-Cas system is not fully clear. One possibility involves autoimmunity, that is, erroneous incorporation of protospacers from the host genome into a CRISPR cassette, resulting in damage to the host chromosome (Stern et al., 2010). Another potentially major source of cost could be the curtailment of horizontal gene transfer (HGT) and hence prevention of genetic novelty acquisition by bacteria and archaea resulting from the CRISPR–Cas activity (Marraffini, 2013; Hatoum-Aslan and Marraffini, 2014). Indeed, it has been shown that multidrug-resistant clinical isolates of enterococci lack CRISPR–Cas, unlike sensitive strains, presumably because having the system hinders resistance plasmid acquisition (Palmer and Gilmore, 2010). The balance between spacer gain and loss could thus be affected by the relative selective pressures exerted, on one hand by viruses and on the other hand by beneficial plasmids (Jiang et al., 2013). Furthermore, CRISPR–Cas can prevent nonparasitic gene acquisition events such as natural transformation with naked DNA (Bikard et al., 2012). It therefore appears that there is a trade-off between active acquired immunity (that is, the constant integration of new spacers and maintenance of large CRISPR arrays) and the ability to acquire new genes and novel functions via HGT. Here we test this hypothesis using available genomic databases and come to the conclusion that on evolutionary timescales, the inhibitory effect of CRISPR–Cas on HGT is undetectable.
Materials and methods
Data
The CRISPR spacer counts were obtained from the September 2013 version of the CRISPRdb (Grissa et al., 2007) and included spacers from all partitions in the genome in question (that is, both chromosome- and plasmid-encoded spacers). The spacer number ranged from 0 to 736.
The updated version of the ATGC database clusters ~60% of proteins and 62% of genomes from RefSeq (as of June 2013) into 269 groups of closely related genomes of bacteria and archaea (Novichkov et al., 2009; Puigbo et al., 2014). Clusters of Orthologous Groups (COGs) shared between genomes in each ATGC were identified (Tatusov et al., 1997; Kristensen et al., 2010), leaving singletons defined as those genes that were not shared with any of the closely related genomes included in the given ATGC. These singletons are most likely to have arisen via recent HGT that has occurred after these closely related organisms have diverged. Only clusters of three or more taxa were used for detection of singletons. For the detection of putative HGT in archaea, the established set of arCOGs (Wolf et al., 2012) (December 2012 release) were employed.
Prophage regions within each genome were identified by PhageFinder (Fouts, 2006). Counts of laterally acquired genes based on unusual dinucleotide composition were obtained from (Popa et al., 2011). Growth temperatures for microbial genomes were downloaded from BacDive (Sohngen et al., 2014) on April 2014.
Statistical analysis
The available data were compiled for 1399 microbial genomes. Decimal logarithms for the fraction of singletons in arCOGs or ATGC COGs, fraction of horizontally transferred genes estimated using the dinucleotide pattern and the fraction of phage-related genes in bacteria were used in the analysis as independent measures of HGT (for correlations between HGT measures, see supplementary Table 1). The number of CRISPR cassette spacers (S) was used in the log10(S+1) form. Growth temperatures were in degree Celsius. The linear model function lm() of the R package was used to estimate the parameters of linear predictors; goodness of fit of alternative models was compared using the ANOVA() function.
Results
Correlating the number of CRISPR spacers and gene acquisition via HGT
There is currently no accurate proxy for CRISPR–Cas activity that can be applied to all microbial genomes. In the absence of a direct measure, spacer count, although not perfect, is the best available genomic proxy for CRISPR activity. It has been established that active CRISPR–Cas systems continuously acquire additional spacers, resulting in longer CRISPR arrays (Tyson and Banfield, 2008). Conversely, spacers can also be quickly deleted (Deveau et al., 2008; Horvath et al., 2008; Tyson and Banfield, 2008), in the absence of selective pressure for maintaining CRIPSR–Cas activity (Weinberger et al., 2012a), because prokaryotic genomes have a bias toward deletions (Kuo and Ochman, 2009), and owing to frequent recombination within repeat loci. Thus, inactive CRISPR arrays will tend to shrink, whereas active ones will expand or at least maintain more or less constant size. Furthermore, even if a CRISPR–Cas system has been recently inactivated in a genome, but this genome still carries a large CRISPR array, it appears more appropriate to view its recent evolutionary history as one where CRISPR–Cas has had an effect.
We therefore used genomic spacer count as a proxy for the CRISPR activity and correlated it with the three available measures of gene acquisition via HGT, namely fraction of prophage genes in bacteria, fraction of singletons in the ATGC COGs and arCOGs, and the fraction of the recently acquired genes inferred on the basis of dinucleotide composition. These relationships show a complex pattern (Tables 1 and 2 and Figure 1), with the magnitude and even the sign of the correlation differing between archaea and bacteria, between CRISPR-positive and CRISPR-negative genomes and between thermophiles and mesophiles. For example, CRISPR-negative bacteria on average encode many fewer prophage-encoded proteins than CRISPR-positive genomes (66.85 vs 114.883, respectively). However, in the CRISPR-positive bacteria, the number of CRISPR spacers negatively correlates with the fraction of prophage genes (Table 2), in agreement with the established role of CRISPR systems in preventing lysogenization (Edgar and Qimron, 2010). In bacteria, the association between spacer number and the the fraction of singletons was nonmonotonic, and surprisingly, the most spacer-rich species also had the highest average fraction of singletons (Figure 1), and the presence of spacers in a genome positively correlated with the fraction of recently acquired genes (both singletons and genes of unusual dinucleotide signature, see Table 2).

The fraction of singleton genes in bacterial genomes binned by CRISPR spacer counts.
In archaeal mesophiles, the fraction of singletons positively correlated with the number of CRISPR spacers (ρ=0.137, N=46), but in the archaeal thermophiles the relationship seems to be reversed (ρ=−0.213, N=65; although both correlation fail to reach statistical significance owing to the small sample size, with P-values of 0.3639 and 0.0887, respectively). In addition, all measures of HGT increase with the genome size, which is a typical dependence for most classes of nonhousekeeping genes (Koonin and Wolf, 2008). In contrast, the number of CRISPR spacers is independent of genome size but has been shown to correlate with the optimal growth temperature (Weinberger et al., 2012b).
Factors affecting gene acquisition
To disentangle the multidimensional network of possibly nonmonotonic relationships, we constructed a linear model to test the predictive power of several variables that could affect the fraction of recently acquired genes. These variables included domain affiliation (Bacteria or Archaea), genome size (the number of protein-coding genes), the growth temperature (either the optimal growth temperature or a binary thermophile vs mesophile classification) and the CRISPR–Cas activity approximated by the number of spacers in CRISPR cassettes (see Supplementary Methods for details). The analysis was started with a model containing all four predictor variables and all their pairwise combinations. The least significant combinations or variables were removed, and the reduced model was compared with the original one using the ANOVA. If the reduced model did not significantly differ from the original full model, the reduction was accepted and the reduced model tested further by removal of the next least significant contributor. Data for 1399 genomes with at least one proxy measure for HGT (fraction of singletons in the context of arCOGs or ATGC COGs, dinucleotide pattern based estimates, number of prophage-related genes in bacteria) available were compiled. Optimal growth temperatures were available for 1034 organisms.
In cases where the growth temperatures were available and included in the model, the number of CRISPR spacers did not contribute significantly to the prediction of the genomic impact of recent HGT, by either method. The optimal predictor for the fraction of singletons included the combined contribution from genome size and growth temperatures, with different offsets for archaea and bacteria (because archaea tend to have many more spacers than bacteria, Figure 2). Altogether, these variables explained ~0.3 of the original variance in the fraction of singletons. When growth temperatures were classified in a binary form (thermophile or mesophile), the number of spacers contributed to the prediction of the number of singletons, along with the genome size, thermophilic status and domain affiliation (Figure 3). Given the strong correlation between the number of CRISPR spacers and the optimal growth temperature (linear correlation coefficient of 0.64), it is likely that the number of spacers is not linked to the extent of HGT directly, but rather serves as a proxy for the temperature. Qualitatively similar results were obtained for the fraction of foreign genes based on dinucleotide pattern and for the fraction of phage-related genes in bacteria (Supplementary Figure 1). Thus, on the scale of evolution of relatively close bacteria and archaea that comprise the ATGC, there is no evidence of a connection between the activity of CRISPR–Cas and HGT.

A predictive model for the fraction of singletons in genomes of archaea and bacteria. Surfaces (top: archaea, bottom: bacteria) indicate the expected fraction of singletons given the optimum growth temperature and genome size.

Predicted and observed fraction of singletons in genomes of archaea and bacteria. Data points correspond to 261 genomes with available optimum growth temperature and the number of singletons. The y=x line is shown.
Discussion
Because CRISPR–Cas systems can prevent plasmid conjugation (Marraffini and Sontheimer, 2008), prophage integration (Edgar and Qimron, 2010) and transformation with naked DNA (Bikard et al., 2012), these immune systems are thought to represent a barrier for HGT that might be detrimental for microbial populations. Observations on human pathogens (Palmer and Gilmore, 2010) as well as a combination of experiments and models (Jiang et al., 2013), have indeed demonstrated that under selective pressure for gene acquisition (for example, exposure to antibiotics, such that acquired resistance is key for survival or replication), CRISPR–Cas systems are often inactivated or lost. Nevertheless, the question of whether indeed there is a general trade-off between possessing efficient protection against selfish genetic elements and access to genetic novelty has not been addressed on a large scale. Here we tested the association between (relatively) recent gene acquisition and CRISPR–Cas activity and observed that overall there is little if any support for the trade-off hypothesis. Indeed, there was no significant negative correlation between CRISPR–Cas activity and the fraction of recently acquired genes (either singletons or genes with unusual dinucleotide composition) in bacteria, whereas the negative correlation observed in archaea seems to merely reflect the anticorrelation between the growth temperature and HGT rate.
The apparent lack of trade-off between CRISPR–Cas activity and gene acquisition via HGT demonstrated in our analysis can be attributed to several nonmutually exclusive evolutionary mechanisms. The simplest explanation is that CRISPR–Cas systems and CRISPR arrays are often themselves mobile so that their presence–absence (inactivity) in any extant genome is not indicative of their longer-term impact. In other words, the relevant events in the evolution of resistance to mobile elements and proclivity for HGT, in which CRISPR–Cas systems have an important role, occur on the population level rather than on the evolutionary scale. Second, in some environments, microbes could experience such high exposure to mobile genetic elements that any CRISPR-mediated resistance to incoming DNA would be ‘a drop in a bucket’. Thus, large CRISPR arrays could indicate genomes that actively acquire new spacers and are under selection to maintain this activity, because they undergo a barrage of selfish DNA elements. In addition, recent evidence has shown that some CRISPR–Cas systems require transcription of the foreign DNA for interference, and thus allow lysogenization, and by inference, at least some forms of HGT, while preventing lytic infection (Goldberg et al., 2014). Third, spacer acquisition in both bacteria and archaea is far from random, with arrays preferentially acquiring new spacers from genomes that contain DNA sequences matching pre-existing spacers, at least partially, a phenomenon known as priming (Datsenko et al., 2012; Swarts et al., 2012; Li et al., 2014). Priming inevitably generates a strong bias toward frequently encountered invasive genetic elements, typically highly infecting viruses, rather than the full spectrum of the exogenous DNA, some of which is potentially beneficial. Indeed, extensive surveys of CRISPR spacers have demonstrated that the majority of matches are to widespread archaeal viruses and bacteriophages, and that multiple spacers potentially matching the same viral genome are often present within the same array (Brodt et al., 2011; Stern et al., 2012). Thus, priming is probably a common mechanism in CRISPR–Cas systems and is likely not only to contribute to more efficient resistance against common viruses but also to limit the novelty-restricting effect of CRISPR–Cas.
What would initially appear like a CRISPR–Cas-mediated barrier to gene transfer in thermophiles and hyperthermophiles, is strongly suggested by our statistical model to reflect a negative association between the growth temperature of an organism and its propensity for taking up novel genes. The barriers to HGT have been intensively studied (Sorek et al., 2007; Wellner et al., 2007; Wellner and Gophna, 2008; Gophna, 2009; Omer et al., 2010; Popa et al., 2011; Naor et al., 2012) but to our knowledge, this work is the first to demonstrate the effect of the growth temperatures on the estimated frequency of gene acquisition. Paradoxically, genomes of extremophilic archaea and bacteria have been known for the large impact of HGT on their evolution, often involving interdomain transfer (Koonin et al., 2001; Gophna et al., 2005). However, most of the HGT events associated with those lineages are ancient ones, often apparently dating back to the emergence of major groups such as Halobacteria, Thermoplasmatales and Archeoglobales (Nelson-Sathi et al., 2012, 2014). In contrast, our present analysis focused on relatively recent gene transfer events that occurred after the divergence of archaeal and bacterial families and genera. Thus, a major influx of foreign genes in the past of a particular lineage does not imply promiscuous HGT at present. Although it appears reasonable to assume that transformation could be anticorrelated with temperature because naked DNA would degrade faster in hotter environments, natural competence has been demonstrated in hyperthermophiles such as Pyrococcus furiosus (Lipscomb et al., 2011) with optimal growth temperatures exceeding 95 °C. In the absence of molecular evidence for the effects of increased temperature, one is left with ecological rationalizations. Higher temperature environments, in particular extreme ones, can be extremely harsh and therefore encompass a lower diversity of microbes (Kemp and Aller, 2004; Miller et al., 2009; López-López et al., 2013) and tighter functional constraints on protein structures (Friedman et al., 2004; Zeldovich et al., 2007; Drake, 2009) both resulting in a smaller gene pool available for HGT.
References
Anderson RE, Brazelton WJ, Baross JA . (2011). Using CRISPRs as a metagenomic tool to identify microbial hosts of a diffuse flow hydrothermal vent viral assemblage. FEMS Microbiol Ecol 77: 120–133.
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315: 1709–1712.
Bikard D, Hatoum-Aslan A, Mucida D, Marraffini LA . (2012). CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe 12: 177–186.
Brodt A, Lurie-Weinberger MN, Gophna U . (2011). CRISPR loci reveal networks of gene exchange in archaea. Biol Direct 6: 65.
Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, Semenova E . (2012). Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun 3: 945.
Denef VJ, Mueller RS, Banfield JF . (2010). AMD biofilms: using model communities to study microbial evolution and ecological complexity in nature. ISME J 4: 599–610.
Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P et al. (2008). Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190: 1390–1400.
Drake JW . (2009). Avoiding dangerous missense: thermophiles display especially low mutation rates. PLoS Genet 5: e1000520.
Edgar R, Qimron U . (2010). The Escherichia coli CRISPR system protects from lambda lysogenization, lysogens, and prophage induction. J Bacteriol 192: 6291–6294.
Fouts DE . (2006). Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res 34: 5839–5851.
Friedman R, Drake JW, Hughes AL . (2004). Genome-wide patterns of nucleotide substitution reveal stringent functional constraints on the protein sequences of thermophiles. Genetics 167: 1507–1512.
Goldberg GW, Jiang W, Bikard D, Marraffini LA . (2014). Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature 514: 633–637.
Gophna U, Doolittle WF, Charlebois RL . (2005). Weighted genome trees: refinements and applications. J Bacteriol 187: 1305–1316.
Gophna U . (2009). Complexity apparently is not a barrier to lateral gene transfers. Microbe 4: 549–553.
Grissa I, Vergnaud G, Pourcel C . (2007). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8: 172.
Gudbergsdottir S, Deng L, Chen Z, Jensen JV, Jensen LR, She Q et al. (2011). Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol Microbiol 79: 35–49.
Hatoum-Aslan A, Marraffini LA . (2014). Impact of CRISPR immunity on the emergence and virulence of bacterial pathogens. Current Opin Microbiol 17: 82–90.
Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA . (2010). Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329: 1355–1358.
Held NL, Herrera A, Cadillo-Quiroz H, Whitaker RJ . (2010). CRISPR associated diversity within a population of Sulfolobus islandicus. PLoS One 5: e12988.
Horvath P, Romero DA, Coute-Monvoisin AC, Richards M, Deveau H, Moineau S et al. (2008). Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol 190: 1401–1412.
Jiang W, Maniv I, Arain F, Wang Y, Levin BR, Marraffini LA . (2013). Dealing with the evolutionary downside of CRISPR immunity: bacteria and beneficial plasmids. PLoS Genet 9: e1003844.
Kemp PF, Aller JY . (2004). Bacterial diversity in aquatic and other environments: what 16 S rDNA libraries can tell us. FEMS Microbiol Ecol 47: 161–177.
Koonin EV, Makarova KS, Aravind L . (2001). Horizontal gene transfer in prokaryotes: quantification and classification. Ann Rev Microbiol 55: 709–742.
Koonin EV, Wolf YI . (2008). Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res 36: 6688–6719.
Kristensen DM, Kannan L, Coleman MK, Wolf YI, Sorokin A, Koonin EV et al. (2010). A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 26: 1481–1487.
Kuo CH, Ochman H . (2009). Deletional bias across the three domains of life. Genome Biol Evol 1: 145–152.
Li M, Wang R, Zhao D, Xiang H . (2014). Adaptation of the Haloarcula hispanica CRISPR-Cas system to a purified virus strictly requires a priming process. Nucleic Acids Res 42: 2483–2492.
Lipscomb GL, Stirrett K, Schut GJ, Yang F, Jenney Jr FE, Scott RA et al. (2011). Natural competence in the hyperthermophilic archaeon Pyrococcus furiosus facilitates genetic manipulation: construction of markerless deletions of genes encoding the two cytoplasmic hydrogenases. Appl Environ Microbiol 77: 2232–2238.
López-López O, Cerdán M, González-Siso M . (2013). Hot spring metagenomics. Life 3: 308–320.
Manica A, Zebec Z, Teichmann D, Schleper C . (2011). In vivo activity of CRISPR-mediated virus defence in a hyperthermophilic archaeon. Mol Microbiol 80: 481–491.
Marraffini LA, Sontheimer EJ . (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322: 1843–1845.
Marraffini LA . (2013). CRISPR-Cas immunity against phages: its effects on the evolution and survival of bacterial pathogens. PLoS Pathogens 9: e1003765.
Miller SR, Strong AL, Jones KL, Ungerer MC . (2009). Bar-coded pyrosequencing reveals shared bacterial community properties along the temperature gradients of two alkaline hot springs in Yellowstone National Park. Appl Environ Microbiol 75: 4565–4572.
Naor A, Lapierre P, Mevarech M, Papke RT, Gophna U . (2012). Low species barriers in halophilic archaea and the formation of recombinant hybrids. Curr Biol 22: 1444–1448.
Nelson-Sathi S, Dagan T, Landan G, Janssen A, Steel M, McInerney JO et al. (2012). Acquisition of 1,000 eubacterial genes physiologically transformed a methanogen at the origin of Haloarchaea. Proc Natl Acad Sci USA 109: 20537–20542.
Nelson-Sathi S, Sousa FL, Roettger M, Lozada-Chavez N, Thiergart T, Janssen A et al. (2014). Origins of major archaeal clades correspond to gene acquisitions from bacteria. Nature 517: 77–80.
Novichkov PS, Ratnere I, Wolf YI, Koonin EV, Dubchak I . (2009). ATGC: a database of orthologous genes from closely related prokaryotic genomes and a research platform for microevolution of prokaryotes. Nucleic Acids Res 37: D448–D454.
Omer S, Kovacs A, Mazor Y, Gophna U . (2010). Integration of a foreign gene into a native complex does not impair fitness in an experimental model of lateral gene transfer. Mol Biol Evol 27: 2441–2445.
Palmer KL, Gilmore MS . (2010). Multidrug-resistant enterococci lack CRISPR-cas. MBio 1: e00227–10.
Popa O, Hazkani-Covo E, Landan G, Martin W, Dagan T . (2011). Directed networks reveal genomic barriers and DNA repair bypasses to lateral gene transfer among prokaryotes. Genome Res 21: 599–609.
Puigbo P, Lobkovsky AE, Kristensen DM, Wolf YI, Koonin EV . (2014). Genomes in turmoil: quantification of genome dynamics in prokaryote supergenomes. BMC Biol 12: 66.
Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B et al. (2011). Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci USA 108: 10098–10103.
Sohngen C, Bunk B, Podstawka A, Gleim D, Overmann J . (2014). BacDive—the Bacterial Diversity Metadatabase. Nucleic Acids Res 42: D592–D599.
Sorek R, Zhu Y, Creevey CJ, Francino MP, Bork P, Rubin EM . (2007). Genome-wide experimental determination of barriers to horizontal gene transfer. Science 318: 1449–1452.
Stern A, Keren L, Wurtzel O, Amitai G, Sorek R . (2010). Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet 26: 335–340.
Stern A, Mick E, Tirosh I, Sagy O, Sorek R . (2012). CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res 22: 1985–1994.
Swarts DC, Mosterd C, van Passel MW, Brouns SJ . (2012). CRISPR interference directs strand specific spacer acquisition. PLoS One 7: e35888.
Tatusov RL, Koonin EV, Lipman DJ . (1997). A genomic perspective on protein families. Science 278: 631–637.
Tyson GW, Banfield JF . (2008). Rapidly evolving CRISPRs implicated in acquired resistance of microorganisms to viruses. Environ Microbiol 10: 200–207.
Weinberger AD, Sun CL, Plucinski MM, Denef VJ, Thomas BC, Horvath P et al. (2012a). Persisting viral sequences shape microbial CRISPR-based immunity. PLoS Comput Biol 8: e1002475.
Weinberger AD, Wolf YI, Lobkovsky AE, Gilmore MS, Koonin EV . (2012b). Viral diversity threshold for adaptive immunity in prokaryotes. MBio 3: e00456–00412.
Wellner A, Lurie MN, Gophna U . (2007). Complexity, connectivity, and duplicability as barriers to lateral gene transfer. Genome Biol 8: R156.
Wellner A, Gophna U . (2008). Neutrality of foreign complex subunits in an experimental model of lateral gene transfer. Mol Biol Evol 25: 1835–1840.
Wolf YI, Makarova KS, Yutin N, Koonin EV . (2012). Updated clusters of orthologous genes for Archaea: a complex ancestor of the Archaea and the byways of horizontal gene transfer. Biol Direct 7: 46.
Zeldovich KB, Berezovsky IN, Shakhnovich EI . (2007). Protein and DNA sequence determinants of thermophilic adaptation. PLoS Comput Biol 3: e5.
Acknowledgements
We thank Kira Makarova and Christine Pourcel for helpful discussions and suggestions, and the eBio IFB platform for bioinformatics support to CRISPRdb. UG is supported by the National Evolutionary Synthesis Center (NESCent). DMK, YIW and EVK are supported by intramural funds of the US Department of Health and Human Services (to the National Library of Medicine). OP is supported by the European Research Council (grant no. 281357).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Gophna, U., Kristensen, D., Wolf, Y. et al. No evidence of inhibition of horizontal gene transfer by CRISPR–Cas on evolutionary timescales. ISME J 9, 2021–2027 (2015). https://doi.org/10.1038/ismej.2015.20
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.20
This article is cited by
-
Phylogenetic Analysis of Anti-CRISPR and Member Addition in the Families
Molecular Biotechnology (2023)
-
CRISPR-Cas systems restrict horizontal gene transfer in Pseudomonas aeruginosa
The ISME Journal (2021)
-
Modeling the ecology of parasitic plasmids
The ISME Journal (2021)
-
The CRISPR-Cas systems were selectively inactivated during evolution of Bacillus cereus group for adaptation to diverse environments
The ISME Journal (2020)
-
Characterization of a SPM-1 metallo-beta-lactamase-producing Pseudomonas aeruginosa by comparative genomics and phenotypic analysis
Scientific Reports (2020)
