
Abstract
Coral and algal holobionts are assemblages of macroorganisms and microorganisms, including viruses, Bacteria, Archaea, protists and fungi. Despite a decade of research, it remains unclear whether these associations are spatial–temporally stable or species-specific. We hypothesized that conflicting interpretations of the data arise from high noise associated with sporadic microbial symbionts overwhelming signatures of stable holobiont members. To test this hypothesis, the bacterial communities associated with three coral species (Acropora rosaria, Acropora hyacinthus and Porites lutea) and two algal guilds (crustose coralline algae and turf algae) from 131 samples were analyzed using a novel statistical approach termed the Abundance-Ubiquity (AU) test. The AU test determines whether a given bacterial species would be present given additional sampling effort (that is, stable) versus those species that are sporadically associated with a sample. Using the AU test, we show that coral and algal holobionts have a high-diversity group of stable symbionts. Stable symbionts are not exclusive to one species of coral or algae. No single bacterial species was ubiquitously associated with one host, showing that there is not strict heredity of the microbiome. In addition to the stable symbionts, there was a low-diversity community of sporadic symbionts whose abundance varied widely across individual holobionts of the same species. Identification of these two symbiont communities supports the holobiont model and calls into question the hologenome theory of evolution.
Similar content being viewed by others


Introduction
The coral holobiont is the assemblage of the coral animal and its symbionts (viruses, Symbiodinium, protozoa, endolithic algae, fungi, Archaea and Bacteria, microfauna) and functions as an ecological unit (Rohwer et al., 2002; Knowlton and Rohwer, 2003). Viruses, mostly phage, are the most abundant and diverse members of the holobiont, followed by Bacteria and Archaea (Kellogg, 2004; Wegley et al., 2007; Marhaver et al., 2008; Vega Thurber et al., 2009). Phage are hypothesized to provide the holobiont with a specific immune system (Barr et al., 2013a,b), in addition to a large reservoir of functional genes (Marhaver et al., 2008; Dinsdale et al., 2008; Vega Thurber et al., 2009; Kelly et al., 2014). The Bacteria and Archaea perform a wide array of metabolic functions that influence the physiology of the holobiont, including cycling of carbon, nitrogen, iron and sulfur (Lesser et al., 2004; Wegley et al., 2007; Beman, 2007; Siboni et al., 2008; Raina et al., 2009; Fiore et al., 2010; Kimes et al., 2010). The microbial symbionts may ward off potentially pathogenic bacteria through niche exclusion and production of antibiotics (Ritchie, 2006; Nissimov et al., 2009; Rypien et al., 2010). Similar to corals, other reef organisms such as sponges (Taylor et al., 2004; Schmitt et al., 2012) and various algal guilds (Lachnit et al., 2009; Barott et al., 2011; Lachnit et al., 2011; Nelson et al., 2013) harbor diverse microbial communities that have been shown or hypothesized to provide ecological services to the holobiont (Lachnit et al., 2009; see review of algal–bacterial associations in Egan et al., 2013). The health of the holobiont is linked to the composition of its viral and microbial constituents, which can become disrupted by natural and anthropogenic stressors, usually leading to blooms of pathogen-related viruses and bacteria (Vega Thurber et al., 2008; Vega Thurber et al., 2009), as well as an increased incidence of diseases (Bourne et al., 2008). However, there are documented cases where stressors such as nitrogen (Siboni et al., 2012) and temperature (Berkelmans and Van Oppen, 2006) may increase microbial symbionts that confer environmentally important physiologies, as well as cases where the bacterial communities return to their original composition after a stressor is removed (Bourne et al., 2008; Sweet et al., 2011).
Even though coral reef holobiont research has been very active for over a decade, the stability of these symbioses over time and space remains contentious. Specifically, do viral and microbial symbionts stably associate with their coral or algal hosts? A survey of the literature is summarized in Table 1 and shows that almost half of the reports find species-specific associations, while the remainder demonstrate environment-driven associations between bacteria and corals. This same question arises for algae, as algal-associated bacterial communities show both species specificity (Lachnit et al., 2009, 2011) and environmentally driven dynamics (Lachnit et al., 2011; Case et al., 2011).
To parameterize dynamical models of the holobiont, the field needs robust statistical tests of membership. Here we characterized >350 000 16S rDNA reads from 131 bacterial communities associated with five hosts: three coral species (Acropora hyacinthus, Acropora rosaria and Porites lutea) and two functional groups of benthic algae (Turf and crustose coralline algae (CCA)). A new statistical approach, the Abundance-Ubiquity (AU) test, was developed to objectively identify members of the bacterial community that were stable or sporadic members of these benthic holobionts. Our results support the hypothesis that coral and algal holobionts harbor both a stable microbiome and a locally variable suite of Bacteria.
Materials and methods
Natural history and sampling design
Coral and algal tissue samples were collected from the six most northerly Line Islands. These islands are remote, span a large biogeochemical gradient and include some of the most pristine reefs in the world (DeMartini et al., 2008; Dinsdale, 2008; Sandin et al., 2008; Barott et al., 2010, 2012). Several of the islands are inhabited, leading to significant changes in the microbial, benthic and fish communities (Dinsdale et al., 2008; McDole et al., 2012; Sandin et al., 2008). Coral and algal tissue samples were collected and preserved, and total DNA was extracted in the same way for all samples in this study.
Sample collection
Coral and algae samples were collected from the following six islands of the Line Islands chain of the Central Pacific: Kingman, Palmyra, and Jarvis (USA) and Teraina, Tabuaeran, and Kiritimati (Republic of Kiribati). Tissue samples were taken from apparently healthy tissue of branching Acropora spp. (A. rosaria, A. hycinthus), P. lutea, CCA (Hydrolithon spp.) and Turf assemblages. A total of 131 samples were collected (Supplementary Table S2). Species-level identification was determined from a combination of field observation and molecular identification using the Internal Transcribed Spacer (ITS) and 18S rDNA. Samples were collected at 5–10 m depth using a hollow punch (0.95 cm diameter) and hammer and placed into individual plastic bags under water. Samples were placed into RNAlater within 1 h of collection and frozen at −20 °C.
DNA extraction and PCR amplification
Coral tissue was removed from the skeleton by air brushing the samples with phosphate-buffered saline and EDTA (10 μM EDTA). Similarly, algal tissue was removed from substrate with the airbrush and PBS+EDTA solution. In all, 500 μl of tissue slurry was centrifuged at 13 000 g for 15 min and resuspended in 500 μl of a lysis buffer (50 mM Tris HCL pH 8.3, Sigma-Aldrich (St Louis, MO, USA); 40 mM EDTA pH 8; 0.75 M sucrose, Sigma-Aldrich). Samples were incubated with lysozyme at a final concentration of 1 mg ml−1 for 60 min at 37 °C. Proteinase K (0.5 mg ml−1 final concentration, Sigma-Aldrich) and sodium dodecyl sulfate (1% final) were added, and samples were incubated overnight at 55 °C and subsequently 70 °C for 10 min. In all, 60 μl 3 M sodium acetate (Sigma-Aldrich), 1 μl 10 mg ml−1 glycogen (Sigma-Aldrich) and 600 μl isopropanol were added, and samples were incubated at −20 °C for 2 h to precipitate DNA. Precipitated DNA centrifuged at 13 000 g at 4 °C, washed with 70% cold Ethanol and resuspended in 567 μl Tris-EDTA.
A CTAB (cetyltrimethylammonium bromide) and subsequent phenol/chloroform extraction method was employed (1% sodium dodecyl sulfate, 0.7 M NaCl and 0.27 mM CTAB (Sigma-Aldrich) solution incubated at 65 °C for 10 min). An equal volume of chloroform was added, mixed vigorously and centrifuged at 13 000 g for 2 min. The aqueous layer was then transferred to clean tube. Phenol:chloroform (Sigma-Aldrich) was then added in equal volume, mixed vigorously and centrifuged at 13 000 g for 2 min. Another chloroform extraction step was carried out to finalize the phenol/chloroform extraction. A 0.7 volume of isopropanol was added to the final aqueous layer and incubated for 2 h. The extract was centrifuged at 4 °C for 15 min at 13 000 g to pellet DNA. DNA was resuspended in 50 μl molecular grade water and stored at −20 °C until ready for amplification via PCR.
Sample preparation for 454 sequencing using barcoded primers
Sample DNA template was loaded onto 96-well plates and prepped for sequencing. Primers were designed using software tools BARCRAWL and BARTAB included the 454 adaptor sequence and a barcode sequence (454_27F: GCCTTGCCAGCCCGCTCAGTCAGAGTTTGATCCTGGCTCAG and 454_338R: GCCTCCCTCGCGCCATCAGxxxxxxxxxxxxCATGCTGCCTCCCGTAGGAGT) (Frank 2009). PCR reactions were performed in triplicate. The PCR amplification protocol is as follows: initial start at 94 °C for 3 min, 35 cycles of 94 °C for 45 s, 50 °C 30 s, 72 °C for 1.5 min, and a final extension of 72 °C for 10 min. Samples were held at 4 °C. The three PCR reactions were pooled, and DNA was quantified with PicoGreen (Life Technologies, Carlsbad, CA, USA). Equal amounts of amplicon (normalized to 240 ng) were pooled in a single tube and cleaned using the MoBio Single Tube PCR Cleanup (Carlsbad, CA, USA). This library was then sequenced using the 454 Titanium platform at the San Diego State University.
Sequencing preparation for host identification
18S rDNA fragment was amplified from the total DNA using a custom Illumina-specific primer (Ill_18S_1A: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAAACCTGGTTGATCCTGCCAGT and Ill_18S_564R: GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGCACCAGACTTGCCCTC). The primer contains the 18S rDNA loci-specific oligo as well as a region for a secondary amplification using Illumina’s barcode indices. The PCR amplification protocol for the primary amplification from total DNA was as follows: 94 °C for 3 min, 30 cycles of 94 °C for 1 min, 55 °C 30 s, 72 °C for 1 min, and a final extension of 72 °C for 10 min holding at 4 °C. Ampure beads were used in order to clean PCR reactions for a secondary amplification with eight cycles in order to apply Illumina’s index barcodes. A secondary cleaning step using Ampure beads was performed, individual libraries were quantified using the PicoGreen dsDNA Assay Kit (Life Technologies) and pooled in equal quantities. The final library was sequenced on the MiSeq at San Diego State University. In order to identify coral species, a coral-specific primer targeting the ITS region (A18S_F: GATCGAACGGTTTAGTGAGG and ITS4: TCCTCCGCTTATTGATATGC) was used to amplify from total DNA (Takabayashi et al., 1998). The PCR protocol was used as performed by Takabayashi et al. (1998) Amplicons were cleaned using Ampure beads and sent for Sanger sequencing at Retrogen (San Diego, CA, USA).
Sequence analysis
Using the Quantitative Insights into Microbial Ecology pipeline (Caporaso et al. 2010), reads were denoised, quality filtered with a cutoff quality score of 25, a minimum and maximum length of 200 and 1000 bp, respectively, no ambiguous bases or mismatches were allowed in the primers and checked for chimeras. Barcodes were used to split sequences into their respective libraries. Operational taxonomic units (OTUs) were generated by picking a seed sequence and clustering at a 97% identity threshold using the UCLUST algorithm (Edgar 2010). Representative OTUs were picked from each cluster to be compared against the SILVA database at an 80% confidence (Quast et al., 2013). OTUs that were classified as chloroplasts were filtered from subsequent analysis, and cyanobacteria were subjected to additional comparison against the SILVA rRNA database to filter any additional chloroplast contamination (minimum alignment length of 151 bp, E-value<103).
In order to identify coral hosts, representative ITS reads were blasted against the NR database. The top hit was used to determine coral species identity. For CCA identification, a representative subsample was taken from each 18S rDNA library and compared against the Silva database as a closed reference using UCLUST. Representative reads were taken from each library, trimmed and aligned using ClustalW, providing phylogenetic distances between reads. Because Turf algae are an assemblage rather than a single species, 18S rDNA libraries for Turf sample identification were clustered into OTUs at a 97% similarity using the SILVA reference database as a closed reference (Quast et al., 2013). OTUs that were assigned to Chlorophyta, Rhodophyceae (excluding CCA) or Phaeophyceae were used as the composition of a given assemblage. In order to determine the range of the composition of turf assemblages, a Bray–Curtis dissimilarity matrix was used to compare differences in assemblage composition. Statistical groupings of turf assemblages were identified using the simprof function from the clustsig R library. The mantel function in R was used to perform mantel tests in order to test for an association between the 18S rDNA dissimlarity of CCA and Turf assemblage composition with respective bacterial community composition. Sequences were submitted to MG-RAST under the study accession number 4663447.3.
All statistical tests were performed in R (version 2.15.3). The vegdist function from the vegan library was used to create Bray–Curtis dissimilarity matrices. Prior to multivariate analysis, a square root transformation was applied, and the data were normalized to relative abundances in order to satisfy assumptions of homogeneity in variance and to account for libraries of differing sequencing depths. The isoMDS function was used to generate nMDS plots of Bray–Curtis dissimilarity matrices. The AU test was implemented as described in Supplementary Appendix S1.
The betadisper function from the vegan library was used to estimate each host group’s associated bacterial community variation using a Bray–Curtis distance matrix. The variation of each community was evaluated by placing the dissimilarity between samples into Euclidean space, making the two-dimensional distances between samples roughly equal the compositional dissimilarity. A group centroid was calculated for each host group, and each sample’s distance to their respective group centroid was calculated. These distances to group centroid were used to test for differences in variability for a given host group.
Results
Characterizing the coral and algal host genotypes
A combination of field observations and molecular marker data was used to identify individual coral and algal genotypes. Coral hosts were identified as P. lutea, A. rosaria and A. hyacinthus using sequences from the ITS region. There was a strong association between the Acropora spp. and island (χ2=15.51; P=0.003). All samples from Jarvis were A. hyacinthus, while all samples from Tabuaeran and Palmyra were A. rosaria; (Supplementary Figure S1; Supplementary Table S2). Both Acropora spp. were sampled from Kingman and Teraina; however, neither were sampled on Kiritimati (the most degraded island) owing to inability to locate any Acropora colonies. P. lutea was collected from every island.
The 18S rDNA locus was used to identify CCA genotypes. All CCA were identified as Hydrolithon sp. Genetic distances between aligned 18S rDNA reads were used to create a dissimilarity matrix, and a mantel test was used to test for an association between the genetic distances and island. No association was found between the CCA 18S rDNAs and sampling location (Mantel R=−0.068; P=0.809), therefore all CCA samples were considered as a single group.
Turf algae are assemblages of different algal species, containing a mixture of juvenile macroalgae, filamentous algae and cyanobacteria. The composition of each Turf assemblage was characterized based on 18S and 16S rDNA reads assigned to Chlorophyta, Phaeophyceae, Rhodophyta and Cyanobacteria. Turf assemblage composition differences were assessed with the Bray–Curtis dissimilarity, and the corresponding dissimilarity matrix was used to test for an association with island. No association was found between Turf assemblage composition and island (Mantel R=−0.018; P=0.584), and therefore, all Turf hosts were considered as a single guild for the subsequent analyses.
In total, we examined five different host groupings: A. hyacinthus, A. rosaria, P. lutea, CCA, and Turf. In total, 8 A. hyacinthus, 16 A. rosaria, 34 P. lutea, 35 CCA and 38 Turf bacterial communities were used in this study. Sample distributions across the islands can be found in Supplementary Table S2. The mean number of post-quality 16S rDNA reads per sample was >1800. The average Shannon diversity (H’±s.d.) of associated bacterial communities were 3.2±0.88, 3.3±0.8, 3.6±0.78, 4.4±0.80 and 4.9±1.4 for A. hyacinthus, A. rosaria, P. lutea, CCA and Turf, respectively (Table 2). The mean number of post-quality filtered 18S rDNA reads per CCA and Turf samples were > 57 000. The average Shannon diversity (±s.d.) of the Eukaryotic community associated with CCA and Turf were 1.0±0.79 and 1.1±0.74, respectively.
Characterization of the variance in bacterial community composition
Bray–Curtis dissimilarity was used to generate a dissimilarity matrix for the bacterial communities associated with the coral species (A. hyacinthus, A. rosaria and P. lutea) and the two algal guilds (CCA and Turf). From this dissimilarity matrix, the first two principle coordinates were plotted to visualize bacterial community composition in Euclidean space (Figure 1a). A spatial median was then calculated for each host type, and group centroids were generated. For each sample, the distance to the respective group centroid was computed, generating a distribution of distances for each host (Figure 1a). These distances represent the variability in the composition of the associated bacterial communities between individuals of each host. A larger median distance-to-centroid indicates higher variation in bacterial communities. The bacterial communities associated with corals were significantly more variable than the algal groups (median distance to centroid±s.d.: 0.42±0.11 versus 0.36±0.05, respectively; Figure 1b; P-values in Supplementary Table S1). Of the coral species, A. rosaria and P. lutea had the same degree of variation in their bacterial community (0.49±0.1 and 0.53±0.15, respectively) and were both higher than A. hyacinthus (0.39±0.09; Figure 1b; Supplementary Table S1).

Bacterial communities associated with coral and algal holobionts. Each host’s associated community was assessed at the OTU level with the Bray–Curtis dissimilarity and those dissimilarities were reduced to principle coordinates, embedding them in Euclidean space (a). The variation of each community was evaluated by measuring the Euclidean distance for each sample associated with a host to the host centroid (b). The bar represents the median distance to centroid and the boxes represent upper and lower 25% quartiles. The whiskers represent minimum and maximum values.
AU test
To identify the OTUs responsible for the community variability identified above, a novel statistical analysis termed the AU test was developed (described in detail in Supplementary Appendix S1). The purpose of this analysis was to address and account for potential incomplete sampling of the bacterial community. That is, despite thousands to tens-of-thousands of observations per sample, there remains a probability that any particular OTU was present but not sampled. The approach taken here compares the ubiquity of an OTU (that is, the fraction of individual samples in which that OTU was observed for a given host) to its relative abundance within each sample. As shown in Figure 2, the ubiquity of an OTU and its relative abundance within a host were strongly correlated for the majority of OTUs; however, there were also OTUs that strongly deviated from this pattern.

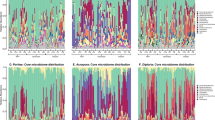
AU method for determining significant deviations in an OTU’s distribution across the samples studied. The AU method was utilized to distinguish between OTUs (97% similar) whose community relative abundance did not vary across each community studied and those that did. OTUs that fall above the 0.01 confidence interval (black line) are found in fewer samples than expected given their mean community relative abundance (that is, OTUs whose distribution in the data set is restricted and are therefore classified as sporadic). Panels a–e represent bacterial communities associated with Acropora hyacinthus (a), Acropora rosaria (b), Porites lutea (c), CCA (d) and Turf (e). Panel f is a mock plot illustrating the different classifications of OTUs based on the AU test. The different color points indicate sporadic (red), stable predicted (blue) and stable observed symbionts (green).
In order to quantify the deviation of an observed AT relationship for an individual OTU and to develop an objective way of identifying the OTUs that exhibit a pattern of high variance in relative abundance, the ubiquity and the pooled relative abundance of OTUs were taken as a pair of test statistics. The distribution of these test statistics was calculated assuming the null hypothesis that an OUT’s relative abundance does not vary across all sampled communities. Figure 2 shows the theoretical expectation curve (blue line), as well as a 0.01 significance curve for these distributions (black line). Any OTU above the significance curve has a probability of <0.01 of achieving ubiquity and abundance under the null assumption. The two groups of OTUs identified by the AU test were labeled as (1) stable symbionts, which were identified by their failure to reject the null hypothesis and therefore display a statistically stable relative abundance across many individuals of a given host group, and (2) sporadic symbionts, which violated the null hypothesis of the AU test, displaying variability in terms of presence/absence and relative abundance between individuals of the same host (Figure 2).
In general, the stable symbionts were rare, where each OTU made up on average 2.6 × 10−4–6.6 × 10−4 of the overall community, and the majority were observed in <50% of the samples (that is, low ubiquity). Therefore, we conditionally classify OTUs that failed to reject the AU test’s null as stable-predicted symbionts. There were also several of OTUs that had a high ubiquity (>50% of samples) and were therefore classified as stable-observed symbionts (the three groups are illustrated in Figure 2f).
Sporadic symbionts are members of the bacterial community that were observed in fewer samples than expected assuming that bacterial associates were stable across time and space. The total proportion of bacteria that were classified as sporadic in the AU test ranged between 4.5% and 7.4% of total OTUs observed with each host (Figure 2; Table 2). The relative abundance of sporadic OTUs was significantly higher than stable OTUs (9.7 × 10−4–8.2 × 10−3; Wilcoxon test; P<0.001; Table 2). The communities of sporadic symbionts were significantly less diverse compared with the stable symbionts across all hosts using the Shannon diversity index (sporadic: 0.794–1.632; stable: 2.177–3.715; Wilcoxon test; P<0.001; Table 2).
Taxonomic identification of stable and sporadic symbionts
The OTUs were taxonomically identified by using the SILVA database as a closed reference and matching using UCLUST (Edgar 2010; Quast et al., 2013). Alphaproteobacteria were the most abundant sporadic symbionts associated with all five hosts. Of these, the majority belonged to the Rhizobiales or Rhodobacterales orders. Turf algae were the exception, with the most abundant OTUs assigned as Rickettsiales.
The most abundant stable symbionts associated with Turf and CCA were Alphaproteobacteria of the orders Rhodobacterales, Rhizobiales and Rhodospirales, represented by the genera Rhodobium and several uncultured/unclassifiable genera (Supplementary Tables S3–S7). Stable symbionts associated with coral also were mostly Alpha and Gammaproteobacteria represented by genera such as Rhodobium, Pseudomonas and Coxiella (Supplementary Tables S3–S7).
There was a single OTU identified as a Propionibacterium sp. that was observed in 100% of the Acropora spp. samples and 95% of the P. lutea samples. This OTU made up on average 3.3% of the community for Acropora spp. and 2.5% on P. lutea. The CCA and turf did not have any OTUs that were observed in >95% of all sampled individuals; however, the most ubiquitously observed OTUs (observed in > 80% of the samples) were all Alphaproteobacteria, mostly belonging to the order Rhodobacterales and Rhizobiales. There was a single Rhodobacterales OTU observed in 92% of turf samples, which on average made 3.1% of the bacterial community. This OTU was also observed in 83% of the CCA samples making up 1.8% of the CCA bacterial communities.
The phylogenetic similarity of stable and sporadic bacteria for each host group was determined using the Bray–Curtis distance at the genus level and a test for significant differences were made using simprof (Supplementary Figure S2). There were five significant groupings at the 0.01 confidence level. A. rosaria and P. lutea stable communities each made up their own grouping while the two algal stable communities made up a significant group on their own (Supplementary Figure S2). The stable community associated with A. hyacinthus formed a group with the algal sporadic communities. Finally, the sporadic communities of coral all formed their own significant group.
Discussion
All macroorganisms associate with microbes, forming ecological assemblages called holobionts (Gordon et al., 2013). Reef corals and algae are some of the best-studied holobionts, but it remains unclear how stable these associations are over time and space (Table 1). Here we developed a statistical method called the AU test to identify different associative patterns of bacterial members of the holobiont. The AU test showed that coral and algal holobionts harbor two different groups of bacterial symbionts: (i) sporadic symbionts, whose abundance varies significantly between individual holobionts of the same species or guild, and (ii) stable symbionts, which are usually found associated with the holobionts of a particular grouping. There were no obligate, exclusive symbionts (that is, a single bacteria species always associated with a single host and not found on other hosts).
Sporadic symbionts
Sporadic symbionts were variably associated with an individual host, but as a group sporadic symbionts were abundant within the holobiont community. The sporadic symbionts are likely the product of either stochastic events or in responses to environmental pressures. In support of the latter hypothesis, other studies have shown that biogeochemical regimes (Siboni et al., 2012; Kelly et al., 2014) or position on the reef (for example, internal reef zones versus reef periphery; Schöttner et al., 2012) influence reef- and organism-associated microbes. Kushmaro and colleagues (Siboni et al., 2012) showed that excess nitrogen led to an enrichment of denitrifying Bacteria and Archaea. Changes in the symbiont members can lead to holobionts better adapted for particular conditions, such as the switching of Symbiodinium clades with depth (Rowan and Knowlton 1995; Toller et al., 2001). Local adaption of the whole reef microbome to local conditions (for example, nutrient regimes) has also been observed in the Line Islands (Kelly et al., 2014). If horizontal acquisition of microbes is the main mechanism for holobiont responses to changing environmental conditions, then adaption is best described as an ecological dynamic. This means that long-term, evolution trajectories are most probably not selected at the level of the holobiont but rather at the species level (that is, swapping of individual members in and out of the holobiont).
Reassessing the ubiquity of a coral symbiont
The Porites astreoides symbiont PA1 (Endozoicomonas sp.) is ubiquitously associated with P. astreoides from Panama to Bermuda (Rohwer et al., 2002). Here we found a Gammaproteobacteria with 99% sequence similarity to PA1, which was found only on P. lutea samples collected from Kingman (10% of all P. lutea samples). Kvennefors et al. (2010) also reported that bacteria similar to PA1 were common on A. hyacinthus and Stylophora pistillata from the Great Barrier Reef, and we observed several additional OTUs that were closely related to PA1 on all the five hosts studied here. Additionally, Bayer et al. (2013) demonstrated that species from the Endozoicomonas genera closely associated with tissue of the coral S. pistillata. Therefore, PA1 and its close relatives appear to associate generally with coral reef macrobes and the abundance of this group is subject to local unknown conditions.
Stable symbionts
Stable bacterial symbionts were relatively rare and highly diverse. These stable symbionts are likely associated with host-constructed niches that are less sensitive to the surrounding environment. For example, Actinobacter and Ralstonia spp. are closely associated with coral gastrodermal cells and endosymbiotic dinoflagellates, suggesting these bacteria occupy specific niches within the macrobial host (Ainsworth et al., 2015). Despite this niche specificity, stable symbionts were not holobiont specific. That is, any holobiont species has a group of stable symbionts; however, these symbionts may also be found on other hosts. This is similar to what has been found for the human and other mammalian microbiomes (Muegge et al., 2011; Human Microbiome Project Consortium, 2012). Additionally, the diversity of these stable symbionts might have a role in the overall community stability (Yachi and Loreau 1999).
Holobionts or hologenomes?
The Holobiont model posits ecological assemblages of symbionts that may change over the lifetime of the macroorganism. The term holobiont was originally coined by Margulis and Fester (1991) and later refined by Rohwer et al. (2002) and Knowlton and Rohwer (2003) to explain the emerging observations about ubiquitous, dynamical symbioses between macrobes and microbes (for example, Symbiodinium and bacteria in corals). The main premises of the holobiont model are: (1) all macroorgansisms form symbioses with microbes, (2) the bionts (viruses, microbes, protists, macrobe and so on) assemble, disassemble and reassemble in different combinations based on ecological dynamics (for example, environmental conditions, migration, predation and so on), and (3) each biont member of the holobiont belongs to a metapopulation with their own evolutionary trajectory. The bionts may be evolutionarily selected based on interactions with each other (that is, coevolution), but they are not obligate symbionts (this work) and they migrate between holobionts and/or other reservoirs (for example, seawater, sediment and so on).
The Hologenome Theory of Evolution was based on the holobiont model and incorporates the exact same premises with one important exception: heredity. To quote from Zilber-Rosenberg and Rosenberg (2008), ‘(1) All animals and plants establish symbiotic relationships with microorganisms. (2) Symbiotic microorganisms are transmitted between generations. (3) The association between host and symbionts affects the fitness of the holobiont within its environment. (4) Variation in the hologenome can be brought about by changes in either the host or the microbiota genomes; under environmental stress, the symbiotic microbial community can change rapidly.’ Therefore, the only strong difference between these two models is the transmission between generations (that is, heredity; Figure 3 compares and contrasts these two models). Here we test the hypothesis that symbionts are transmitted by looking across populations of coral and algal holobionts. If bacteria are transmitted between generations, then these bacteria should be ubiquitous and have strong phylosymbiotic signals (Brucker and Bordenstein, 2013). The analysis here shows that this is not true; the stable bacterial symbionts are neither ubiquitous nor specific to one host. Ainsworth et al. (2015) came to similar conclusions sans an AU-like test. Similar to Symbiodinium, the vast majority of bacterial symbionts are moving in and out of the holobiont. This is also similar to what has been observed in the human microbiome (Human Microbiome Project Consortium, 2012), and the AU test described here can be used to assess the generalization of this phenomenon across all holobionts.

Comparison of the Holobiont model (Rohwer et al. 2002; Knowlton and Rohwer, 2003) and Hologenomic Theory of Evolution (Rosenberg et al., 2007). The primary difference between the two hypotheses is the importance of heredity. For the illustration, the coral animal is represented by hexagons, the colored circles represent Symbiodinium and oval shapes represent Bacteria. In actuality, the symbionts also include viruses, Archaea, fungi, protists, other macrobes and so on. In the Holobiont model, (a) heritability is not required and the relationships among the bionts are best described by ecological dynamics. Membership in the holobiont may be stable or sporadic as predicted by the AU test (blue ovals in figure) or obligate (for example, plastids or mitochondria). Stable and sporadic symbionts are recruited from other holobionts or from the environment (different color ovals). Changing environmental conditions lead to symbiont switching. Symbionts remain independent entities, may be found outside the holobiont and are sometimes absent from any individual holoboint of the same species (that is, not ubiquitous, similar to PA1). Even in a stable environment, members of the holobiont may be replaced by bionts with similar functions. (b) The Hologenome is evolutionary and requires heredity, otherwise it incorporates the same premises laid forth under the Holobiont model and is indistinguishable.
Based on the observations presented here and in Table 1, we propose that the holobiont model best describes what has been observed for macrobe–microbe associations. One place where additional clarity is needed is when two bionts become linked in an obligate manner (for example, Ochman and Moran 2001; Moran et al., 2008; and Brucker and Bordenstein 2013; Bordeinstein and Theis, 2015). Endosymbiotic theory and the subsequent models for understanding this type of symbiosis are established (Margulis and Fester, 1991). To differentiate between obligate symbioses and the more flexible, holobiont-type found in corals, algae and human, we propose that there is a bifurcation called aboluta iunctio for ‘absolute linkage’ where two symbionts become one, indivisible biont (based on some to be determined by probability). The attraction of incorporating aboluta iunctio into models of symbioses is that it would be both mathematically tractable and translatable into laboratory and field studies.
What to do with the hologenome?
Despite the eloquent defense of the term by Rosenberg et al., 2007, the term hologenome has become divisive and the older term metagenome should be used to describe the collective genetic material of a particular sample (holobiont or otherwise). As for the Hologenome Theory of Evolution, without clear evolutionary mechanisms, including heredity, this is a misnomer. It might be useful to develop the theory as a null model for multilevel selection, but this will require more quantitative precision than has been forthcoming. Statements that hologenomes are integrated such that they are free from conflict (Rosenberg et al., 2007; Rosenberg and Zilber-Rosenberg, 2013) has eroded any serious consideration by evolutionary theorists. This assertion is clearly not true; symbiont members of the assemblages frequently kill or weaken the holobiont in corals (Kline et al., 2006; Vega Thurber et al., 2008; Vega Thurber et al., 2009). Further, the very name hologenome implies a gene-centric approach reminiscent of the selfish gene. And while it is possible to measure the outcomes of evolutionary processes at the genetic level, this information is only relevant within the context of its environment (for example, in a cell). Bacteria, Symbiodinium, corals and the other bionts of the holobiont have remained separate entities despite close association for >200 million years (Timmis et al., 2004; Life and Death of Corals Reefs, chapter 5) and it is the bionts, not just their genetic material, that determines selectable phenotypes (Grafen and Ridley 2007).
Conclusions
The results here support the holobiont model where the microbial and macrobial members have individual evolutionary trajectories. Although there are stable associations between the bacteria and macrobes, these associations are not exclusive and therefore not heritable as a unit. Our emerging view is that coral and algal holobionts assemble and then the environment selects for membership. As the environment changes, there is reassembling and the acquisition of new members. This is similar to the adaptive bleaching model proposed by Buddemeier and Fautin (1993). As with the Symbiodinium, our analysis suggests that the stable bacterial symbionts are found on multiple hosts. The fact that the individual elements of the holobiont have remained separate for several hundreds of millions of years also suggests that the unit of selection is at the individual biont level. Until compelling contradictory evidence is presented, the gene-centric proposal of the hologenome is subject to all of the criticisms of the selfish gene hypothesis, as well as the challenges presented by this study (that is, heritability). At this stage, the evidence points toward holobionts. However, to really understand the complex dynamics in these systems, there is a need for strong mathematical models, extensive sampling of naturally occurring holobionts and statistics, such as the AU test.
References
Ainsworth T, Krause L, Bridge T, Torda G, Raina J-B, Zakrzewski M et al. (2015). The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J 9: 2261–2274.
Aires T, Moalic Y, Serrao EA, Arnaud-Haond S . (2015). Hologenome theory supported by cooccurrence networks of species-specific bacterial communities in siphonous algae (Caulerpa. FEMS Microbiol Ecol 91.
Barr JJ, Youle M, Rohwer F . (2013a). Innate and acquired bacteriophage-mediated immunity. Bacteriophage 3: 10771–10776.
Barr JJ, Auro R, Furlan M, Whiteson KL, Erb ML, Pogliano J et al. (2013b). Bacteriophage adhering to mucus provide a non–host-derived immunity. Proc Natl Acad Sci 110: 10771–10776.
Barott KL, Caselle JE, Dinsdale EA, Friedlander AM, Maragos JE, Obura D et al. (2010). The lagoon at Caroline/Millennium Atoll, Republic of Kiribati: natural history of a nearly pristine ecosystem. PLoS One 5: e10950.
Barott KL, Rodriguez‐Brito B, Janouškovec J, Marhaver KL, Smith JE, Keeling P et al. (2011). Microbial diversity associated with four functional groups of benthic reef algae and the reef‐building coral Montastraea annularis. Environ Microbiol 13: 1192–1204.
Barott KL, Williams GJ, Vermeij MJ, Harris J, Smith JE, Rohwer FL et al. (2012). Natural history of coral− algae competition across a gradient of human activity in the Line Islands. Institute for Biodiversity and Ecosystem Dynamics (IBED).
Bayer T, Neave MJ, Alsheikh-Hussain A, Aranda M, Yum LK, Mincer T et al. (2013). The microbiome of the Red Sea coral Stylophora pistillata is dominated by tissue-associated Endozoicomonas bacteria. Appl Environ Microbiol 79: 4759–4762.
Beman JM, Roberts KJ, Wegley L, Rohwer F, Francis CA . (2007). Distribution and diversity of archaeal ammonia monooxygenase genes associated with corals. Appl Environ Microbiol 73: 5642–5647.
Berkelmans R, Van Oppen MJ . (2006). The role of zooxanthellae in the thermal tolerance of corals: a ‘nugget of hope’ for coral reefs in an era of climate change. Proc R Soc London B Biol Sci 273: 2305–2312.
Birkeland C . (1997) Life and Death of Coral Reefs. Springer Science & Business Media.
Bourne D, Iida Y, Uthicke S, Smith-Keune C . (2008). Changes in coral-associated microbial communities during a bleaching event. ISME J 2: 350–363.
Bourne DG, Munn CB . (2005). Diversity of bacteria associated with the coral Pocillopora damicornis from the Great Barrier Reef. Environ Microbiol 7: 1162–1174.
Bordenstein SR, Theis KR . (2015). Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PLoS Biol 13: e1002226.
Brucker RM, Bordenstein SR . (2013). The hologenomic basis of speciation: gut bacteria cause hybrid lethality in the genus. Nasonia Sci 341: 667–669.
Buddemeier RW, Fautin DG . (1993). Coral bleaching as an adaptive mechanism. Bioscience 43: 320–326.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Carlos C, Torres TT, Ottoboni LM . (2013). Bacterial communities and species-specific associations with the mucus of Brazilian coral species. Sci Rep 3: 1624.
Case RJ, Longford SR, Campbell AH, Low A, Tujula N, Steinberg PD et al. (2011). Temperature induced bacterial virulence and bleaching disease in a chemically defended marine macroalga. Environ Microbiol 13: 529–537.
Ceh J, Van Keulen M, Bourne DG . (2011). Coral-associated bacterial communities on Ningaloo Reef, Western Australia. FEMS Microbiol Ecol 75: 134–144.
Chen CP, Tseng CH, Chen CA, Tang SL . (2011). The dynamics of microbial partnerships in the coral Isopora palifera. ISME J 5: 728–740.
Chiu JM, Li S, Li A, Po B, Zhang R, Shin PK et al. (2012). Bacteria associated with skeletal tissue growth anomalies in the coral Platygyra carnosus. FEMS Microbiol Ecol 79: 380–391.
Daniels CA, Zeifman A, Heym K, Ritchie KB, Watson CA, Berzins I et al. (2011). Spatial heterogeneity of bacterial communities in the mucus of Montastraea annularis. Ma. Ecol Prog Ser 426: 29–40.
DeMartini EE, Friedlander AM, Sandin SA, Sala E . (2008). Differences in fish-assemblage structure between fished and unfished atolls in the northern Line Islands, central Pacific. Mar Ecol Progr Ser 365: 199–215.
Dinsdale EA, Pantos O, Smriga S, Edwards RA, Angly F, Wegley L et al. (2008). Microbial ecology of four coral atolls in the Northern Line Islands. PloS One 3: e1584.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Egan S, Harder T, Burke C, Steinberg P, Kjelleberg S, Thomas T . (2013). The seaweed holobiont: understanding seaweed–bacteria interactions. FEMS Microbiol Rev 37: 462–476.
Fiore CL, Jarett JK, Olson ND, Lesser MP . (2010). Nitrogen fixation and nitrogen transformations in marine symbioses. Trends Microbiol 18: 455–463.
Frank DN . (2009). BARCRAWL and BARTAB: software tools for the design and implementation of barcoded primers for highly multiplexed DNA sequencing. BMC Bioinform 10: 362.
Frias-Lopez J, Zerkle AL, Bonheyo GT, Fouke BW . (2002). Partitioning of bacterial communities between seawater and healthy, black band diseased and dead coral surfaces. Appl Environ Microbiol 68: 2214–2228.
Gordon J, Knowlton N, Relman DA, Rohwer F, Youle M . (2013). Superorganisms and holobionts. Microbe 8: 152–153.
Grafen A, Ridley M . (2007) Richard Dawkins: How a Scientist Changed the way we Think: Reflections by Scientists, Writers, and Philosophers. Oxford University Press.
Guppy R, Bythell JC . (2006). Environmental effects on bacterial diversity in the surface mucus layer of the reef coral Montastraea faveolata. Mar Ecol Prog Ser 328: 133–142.
Hong MJ, Yu YT, Chen CA, Chiang PW, Tang SL . (2009). Influence of species specificity and other factors on bacteria associated with the coral Stylophora pistillata in Taiwan. Appl Environ Microbiol 75: 7797–7806.
Human Microbiome Project Consortium. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214.
Kellogg C . (2004). Tropical Archaea: diversity associated with the surface microlayer of corals. Mar Ecol Prog Ser 273: 81−88.
Kelly LW, Williams GJ, Barott KL, Carlson CA, Dinsdale EA, Edwards RA et al. (2014). Local genomic adaptation of coral reef-associated microbiomes to gradients of natural variability and anthropogenic stressors. Proc Natl Acad Sci USA 111: 10227–10232.
Kimes NE, Johnson WR, Torralba M, Nelson KE, Weil E, Morris PJ . (2013). The Montastraea faveolata microbiome: ecological and temporal influences on a Caribbean reef‐building coral in decline. Environ Microbiol 15: 2082–2094.
Kimes NE, Van NostrKimes NE, Van Nostrand JD, Weil E, Zhou J, Morris PJ . (2010). Microbial functional structure of Montastraea faveolata, an important Caribbean reef‐building coral, differs between healthy and yellow‐band diseased colonies. Environ Microbiol 12: 541–556.
Klaus JS, Frias-Lopez J, Bonheyo GT, Heikoop JM, Fouke BW . (2005). Bacterial communities inhabiting the healthy tissues of two Caribbean reef corals: interspecific and spatial variation. Coral Reefs 24: 129–137.
Klaus JS, Janse I, Heikoop JM, Sanford RA, Fouke BW . (2007). Coral microbial communities, zooxanthellae and mucus along gradients of seawater depth and coastal pollution. Environ Microbiol 9: 1291–1305.
Kline DI, Kuntz NM, Breitbart M, Knowlton N, Rohwer F . (2006). Role of elevated organic carbon levels and microbial activity in coral mortality. Mar Ecol Progr Ser 314: 119–125.
Knowlton N, Rohwer F . (2003). Multispecies microbial mutualisms on coral reefs: the host as a habitat. Am Nat 162: S51–S62.
Kvennefors ECE, Sampayo E, Ridgway T, Barnes AC, Hoegh-Guldberg O . (2010). Bacterial communities of two ubiquitous Great Barrier Reef corals reveals both site-and species-specificity of common bacterial associates. PLoS One 5: e10401.
Lachnit T, Meske D, Wahl M, Harder T, Schmitz R . (2011). Epibacterial community patterns on marine macroalgae are host‐specific but temporally variable. Environ Microbiol 13: 655–665.
Lachnit T, Wahl M, Harder T . (2009). Isolated thallus-associated compounds from the macroalga Fucus vesiculosus mediate bacterial surface colonization in the field similar to that on the natural alga. Biofouling 26: 247–255.
Lee OO, Yang J, Bougouffa S, Wang Y, Batang Z, Tian R et al. (2012). Spatial and species variations in bacterial communities associated with corals from the Red Sea as revealed by pyrosequencing. Appl Environ Microbiol 78: 7173–7184.
Lema KA, Willis BL, Bourne DG . (2014). Amplicon pyrosequencing reveals spatial and temporal consistency in diazotroph assemblages of the Acropora millepora microbiome. Environ Microbiol 16: 3345–3359.
Lesser MP, Mazel CH, Gorbunov MY, Falkowski PG . (2004). Discovery of symbiotic nitrogen-fixing cyanobacteria in corals. Science 305: 997–1000.
Li J, Chen Q, Zhang S, Huang H, Yang J, Tian XP et al. (2013). Highly heterogeneous bacterial communities associated with the South China Sea reef corals Porites lutea Galaxea fascicularis and Acropora millepora. PLoS One 8: e71301.
Lins-de-Barros MM, Vieira RP, Cardoso AM, Monteiro VA, Turque AS, Silveira CB et al. (2010). Archaea, Bacteria, and algal plastids associated with the reef-building corals Siderastrea stellata and Mussismilia hispida from Buzios, South Atlantic Ocean, Brazil. Microb Ecol 59: 523–532.
Littman RA, Willis BL, Pfeffer C, Bourne DG . (2009). Diversities of coral-associated bacteria differ with location, but not species, for three acroporid corals on the Great Barrier Reef. FEMS Microbiol Ecol 68: 152–163.
Margulis L, Fester R . (1991) Symbiosis as a Source of Evolutionary Innovation: Speciation and Morphogenesis. MIT Press.
Marhaver KL, Edwards RA, Rohwer F . (2008). Viral communities associated with healthy and bleaching corals. Environ Microbiol 10: 2277–2286.
McDole T, Nulton J, Barott KL, Felts B, Hand C, Hatay M et al. (2012). Assessing coral reefs on a Pacific-wide scale using the microbialization score. PloS One 7: e43233.
McKew BA, Dumbrell AJ, Daud SD, Hepburn L, Thorpe E, Mogensen L et al. (2012). Characterization of geographically distinct bacterial communities associated with coral mucus produced by Acropora spp. and Porites spp. Appl Environ Microbiol 78: 5229–5237.
Moran NA, McCutcheon JP, Nakabachi A . (2008). Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet 42: 165–190.
Morrow KM, Moss AG, Chadwick NE, Liles MR . (2012). Bacterial associates of two Caribbean coral species reveal species-specific distribution and geographic variability. Appl Environ Microbiol 78: 6438–6449.
Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, Fontana L et al. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332: 970–974.
Nelson CE, Goldberg SJ, Kelly LW, Haas AF, Smith JE, Rohwer F et al. (2013). Coral and macroalgal exudates vary in neutral sugar composition and differentially enrich reef bacterioplankton lineages. ISME J 7: 962–979.
Nissimov J, Rosenberg E, Munn CB . (2009). Antimicrobial properties of resident coral mucus bacteria of Oculina patagonica. FEMS Microbiol Lett 292: 210–215.
Ochman H, Moran NA . (2001). Genes lost and genes found: evolution of bacterial pathogenesis and symbiosis. Science 292: 1096–1099.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41: D590–D596.
Raina JB, Tapiolas D, Willis BL, Bourne DG . (2009). Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl Environ Microbiol 75: 3492–3501.
Reshef L, Koren O, Loya Y, Zilber‐Rosenberg I, Rosenberg E . (2006). The coral probiotic hypothesis. Environ Microbiol 8: 2068–2073.
Ritchie KB . (2006). Regulation of microbial populations by coral surface mucus and mucus-associated bacteria. Mar Ecol Progr Ser 322: 1–14.
Rohwer F, Seguritan V, Azam F, Knowlton N . (2002). Diversity and distribution of coral-associated bacteria. Mar Ecol Progr Ser 243: 1–10.
Rosenberg E, Koren O, Reshef L, Efrony R, Zilber-Rosenberg I . (2007). The role of microorganisms in coral health, disease and evolution. Nat Rev Microbiol 5: 355–362.
Rosenberg E, Zilber-Rosenberg I . (2013) The Hologenome Concept: Human, Animal and Plant Microbiota. Springer: Berlin, Heidelberg, Germany.
Rowan R, Knowlton N . (1995). Intraspecific diversity and ecological zonation in coral-algal symbiosis. Proc Natl Acad Sci 92: 2850–2853.
Rypien KL, Ward JR, Azam F . (2010). Antagonistic interactions among coral‐associated bacteria. Environ Microbiol 12: 28–39.
Sandin SA, Smith JE, Demartini EE, Dinsdale EA, Donner SD, Friedlander AM et al. (2008). Baselines and degradation of coral reefs in the Northern Line Islands. PloS One 3: e1548.
Schmitt S, Tsai P, Bell J, Fromont J, Ilan M, Lindquist N et al. (2012). Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J 6: 564–576.
Schöttner S, Wild C, Hoffmann F, Boetius A, Ramette A . (2012). Spatial scales of bacterial diversity in cold-water coral reef ecosystems. PloS One 7: e32093.
Siboni N, Ben‐Dov E, Sivan A, Kushmaro A . (2008). Global distribution and diversity of coral‐associated Archaea and their possible role in the coral holobiont nitrogen cycle. Environ Microbiol 10: 2979–2990.
Siboni N, Ben-Dov E, Sivan A, Kushmaro A . (2012). Geographic specific coral-associated ammonia-oxidizing archaea in the northern Gulf of Eilat (Red Sea). Microb Ecol 64: 18–24.
Sunagawa S, Woodley CM, Medina M . (2010). Threatened corals provide underexplored microbial habitats. PLoS One 5: e9554.
Sweet MJ, Croquer A, Bythell JC . (2011). Bacterial assemblages differ between compartments within the coral holobiont. Coral Reefs 30: 39–52.
Takabayashi M, Carter DA, Loh WKW, Hoegh-Guldberg O . (1998). A coral-specific primer for PCR amplification of the internal transcribed spacer region in ribosomal DNA. Mol Ecol 7: 928–930.
Taylor MW, Schupp PJ, Dahllöf I, Kjelleberg S, Steinberg PD . (2004). Host specificity in marine sponge‐associated bacteria, and potential implications for marine microbial diversity. Environ Microbiol 6: 121–130.
Toller WW, Rowan R, Knowlton N . (2001). Zooxanthellae of the Montastraea annularis species complex: patterns of distribution of four taxa of Symbiodinium on different reefs and across depths. Biol Bull 201: 348–359.
Tout J, Jeffries TC, Webster NS, Stocker R, Ralph PJ, Seymour JR . (2014). Variability in microbial community composition and function between different niches within a coral reef. Microb Ecol 67: 540–552.
Timmis JN, Ayliffe MA, Huang CY, Martin W . (2004). Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat Rev Genet 5: 123–135.
Tremblay P, Weinbauer MG, Rottier C, Guérardel Y, Nozais C, Ferrier-Pagès C . (2011). Mucus composition and bacterial communities associated with the tissue and skeleton of three scleractinian corals maintained under culture conditions. Mar Biol Assoc UK 91: 649.
Vega Thurber RL, Barott KL, Hall D, Liu H, Rodriguez-Mueller B, Desnues C et al. (2008). Metagenomic analysis indicates that stressors induce production of herpes-like viruses in the coral Porites compressa. Proc Natl Acad Sci USA 105: 18413–18418.
Vega Thurber R, Willner‐Hall D, Rodriguez‐Mueller B, Desnues C, Edwards RA, Angly F et al. (2009). Metagenomic analysis of stressed coral holobionts. Environ Microbiol 11: 2148–2163.
Wegley L, Edwards R, Rodriguez‐Brito B, Liu H, Rohwer F . (2007). Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ Microbiol 9: 2707–2719.
Yachi S, Loreau M . (1999). Biodiversity and ecosystem productivity in a fluctuating environment: the insurance hypothesis. Proc Natl Acad Sci 96: 1463–1468.
Yang S, Sun W, Zhang F, Li Z . (2013). Phylogenetically diverse denitrifying and ammonia-oxidizing bacteria in corals Alcyonium gracillimum and Tubastraea coccinea. Mar Biotechnol 15: 540–551.
Zilber-Rosenberg I, Rosenberg E . (2008). Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution. FEMS Microbiol Rev 32: 723–735.
Acknowledgements
We thank Peter Salamon, Barbara Bailey and Ben Felts from the SDSU Biomath group, as well as Linda Wegley, Nate Robinett and Emma Ransome, for insightful discussion. Thank you to Dana Berg-Lyons and Rob Knight for support with amplicon library preparations and providing the 16S rDNA barcode primers. Jennifer Smith and Gareth Williams helped identify Acropora spp. and Porites spp. in the field. We also thank all of the members of the Line Islands 2010 cruise for their work in the field. Samples were collected during a research expeditions to the Northern Line Islands funded by the Moore Family Foundation, the Hawaii Undersea Research Laboratory of the Coral Reef Conservation Program (a program of the National Oceanic and Atmospheric Administration (NOAA)) and several private donors. This work was carried out under research permits from the Palmyra Atoll National Wildlife Refuge, US Fish and Wildlife Service, and the Environment and Conservation Division of the Republic of Kiribati (12533-07021 and 12533-10010). This research was sponsored by National Science Foundation Awards OISE/IIA-1243541 (to FR), GBMF MMI Investigator award (Moore-3781; to FR) and a Canadian Institute for Advanced Research Integrated Microbial Biodiversity Program Fellowship 141679 (to FR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Hester, E., Barott, K., Nulton, J. et al. Stable and sporadic symbiotic communities of coral and algal holobionts. ISME J 10, 1157–1169 (2016). https://doi.org/10.1038/ismej.2015.190
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.190
This article is cited by
-
A coral-associated actinobacterium mitigates coral bleaching under heat stress
Environmental Microbiome (2023)
-
The microbiome of the endosymbiotic Symbiodiniaceae in corals exposed to thermal stress
Hydrobiologia (2023)
-
Diversity of the Pacific Ocean coral reef microbiome
Nature Communications (2023)
-
Microbiomes of the Sydney Rock Oyster are acquired through both vertical and horizontal transmission
Animal Microbiome (2022)
-
Microbiota mediated plasticity promotes thermal adaptation in the sea anemone Nematostella vectensis
Nature Communications (2022)
