
Abstract
The colonization pattern of intestinal microbiota during childhood may impact health later in life, but children older than 1 year are poorly studied. We followed healthy children aged 1–4 years (n=28) for up to 12 months, during which a synbiotic intervention and occasional antibiotics intake occurred, and compared them with adults from the same region. Microbiota was quantified with the HITChip phylogenetic microarray and analyzed with linear mixed effects model and other statistical approaches. Synbiotic administration increased the stability of Actinobacteria and antibiotics decreased Clostridium cluster XIVa abundance. Bacterial diversity did not increase in 1- to 5-year-old children and remained significantly lower than in adults. Actinobacteria, Bacilli and Clostridium cluster IV retained child-like abundances, whereas some other groups were converting to adult-like profiles. Microbiota stability increased, with Bacteroidetes being the main contributor. The common core of microbiota in children increased with age from 18 to 25 highly abundant genus-level taxa, including several butyrate-producing organisms, and developed toward an adult-like composition. In conclusion, intestinal microbiota is not established before 5 years of age and diversity, core microbiota and different taxa are still developing toward adult-type configuration. Discordant development patterns of bacterial phyla may reflect physiological development steps in children.
Similar content being viewed by others
Introduction
Numerous studies have identified associations between aberrant composition of microbiota in children and diseases including atopic diseases, autism, obesity, Crohn's disease, celiac disease and food allergies (Kalliomäki et al., 2008; Cucchiara et al., 2009; Frank et al., 2011; Cheng et al., 2013; Azad et al., 2014; Nylund et al., 2015; Toh and Allen-Vercoe, 2015). Decreased diversity and richness of microbiota has been linked to several disorders (Turnbaugh and Gordon, 2009; Verdam et al., 2013; Nylund et al., 2015). For instance, the diversity of microbial community and the abundance of butyrate-producing bacteria correlate negatively with the severity of atopic eczema in young children (Nylund et al., 2015). Furthermore, reduced diversity and Bacteroidetes/Firmicutes ratio is associated with obesity (Ley et al., 2006; Turnbaugh and Gordon, 2009; Verdam et al., 2013). Importantly, aberrancies in early-life microbiota development may impact the health status also later in life (Rautava et al., 2012; Nylund et al., 2014). For example, low levels of bifidobacteria in infancy may predispose to overweight at the age of 7 years (Kalliomäki et al., 2008). Understanding of the microbiota development during childhood is essential in identifying windows of opportunity to influence the microbiota development.
The major colonization of gut microbiota begins during birth, when infants are exposed to extra-uterine environment. The microbiota development is affected by a number of factors including genetics, mode of delivery and gestational age, antibiotic usage, breast feeding (BF)/formula feeding and long-term diet after weaning (Penders et al., 2006; Grönlund et al., 2007; Dominguez-Bello et al., 2010; Ladirat et al., 2013; Azad et al., 2014; Nylund et al., 2014; Olivares et al., 2014; Voreades et al., 2014). During the colonization process, facultative anaerobic bacteria, such as Enterobacteria, are gradually replaced by anaerobic bacteria, such as the genera Bifidobacterium, Clostridium and Bacteroides (Weber and Polanco, 2012; Matamoros et al., 2013; Arrieta et al., 2014). The major diversification of microbiota is associated with the introduction of solid foods at 4–6 months of age (Fallani et al., 2011; Koenig et al., 2011; Bergström et al., 2014).
When compared with adults or older children, the infant microbiota has low diversity and the microbiota structure is generally unstable and highly dynamic (Arrieta et al., 2014). In contrast, the gut microbiota of healthy adults is individual specific and relatively stable in time (Claesson et al., 2011; Jalanka-Tuovinen et al., 2011; Rajilić-Stojanović et al., 2013). Two main phyla, Firmicutes and Bacteroidetes, constitute over 90% of the microbiota in healthy adults, followed by Actinobacteria, Proteobacteria and Verrucomicrobia (Claesson et al., 2011; Jalanka-Tuovinen et al., 2011; Ringel-Kulka et al., 2013; Voreades et al., 2014). Although numerous studies have investigated the composition and dynamics of the intestinal microbiota in infants and adults, the gut microbiota of toddler and pre-school children remains poorly characterized, representing a missing link in the long-term dynamics. Several studies have reported that the microbiota starts to resemble adult-like microbiota within 2–3 years after birth (Koenig et al., 2011; Yatsunenko et al., 2012; Bergström et al., 2014), but differences in the bacterial community structure and diversity still exist between toddlers and adults (Ringel-Kulka et al., 2013). It is still not known at what age the individual-specific adult-like microbiota profile is established (Koenig et al., 2011; Bergström et al., 2014; Nylund et al., 2014). Therefore, the dynamics of microbiota development as well as the ‘normal course’ of microbiota development after infancy are still poorly understood.
The aim of this longitudinal study was to follow the development of microbiota during 1 year in order to investigate whether children’s microbiota convert to adult-type composition and diversity in young children at 1–5 years of age, and to evaluate the effect of a synbiotic intervention on fecal microbiota.
Materials and methods
Subjects
Fecal samples were collected from healthy subjects, including 28 children (12–48 months) and 23 adults (21–60 years of age) from North Carolina, USA. The criteria for selecting the participants are described in detail in the previous cross-sectional study (Ringel-Kulka et al., 2013). The demographic characteristics for both children and adults are compiled in Supplementary Table 1. The studied adult population is culturally relatively homogeneous, with concerns for their fairly similar diet. All children participating in the study were attending daycare centers and therefore during working days received meals that are compiled according to official nutritional requirements for the age group.
In this follow-up study, stool samples from all children were sampled at the baseline and continually during 1 year (see Supplementary Figure 1 for experimental design). After baseline sampling, the children entered a synbiotic intervention trial for 4 months (Probiotics: Bifidobacterium animalis subsp. lactis BB-12, Streptococcus thermophilus and Lactobacillus bulgaricus and inulin, Placebo: acidified milk) described in detail previously (Ringel-Kulka et al., 2015). The children had no history of treatment with antibiotics within the previous 4 weeks, and no intentional consumption of probiotics within 2 weeks before the intervention. Fecal samples were obtained at baseline, at end of the intervention, and 2 and 8 months after the intervention. At the end of the intervention, fecal samples were obtained from 25 children, 12 samples from the synbiotic intervention group and 13 from the placebo group. Furthermore, 20 subjects provided follow-up samples 2 months after the intervention and 10 subjects provided follow-up samples 8 months after the intervention (Supplementary Table 1a). During the intervention and follow-up time periods, 11 subjects received short-term antibiotics treatment for acute infections, such as ear infections. For each child, a body mass index (BMI) scaled according to the BMI distribution in the respective age and gender categories, the Z-score BMI (zBMI), was determined. All recorded data are compiled in Supplementary Table 1a.
Ethical considerations
The study was approved by the University of North Carolina (UNC) Internal Review Board. All adult subjects and parents/guardians of the children provided written consent before study participation.
Fecal samples and DNA extraction
The sample collection and preparation has been described previously (Carroll et al., 2011; Ringel-Kulka et al., 2013). In short, childrens' fecal samples were collected at home and returned to the daycare center following previously described instructions (Ringel-Kulka et al., 2015). The adult fecal samples were collected at a visit at UNC clinic or at home. The samples collected at home were transferred to daycare center or UNC clinic within 12 h from defecation in a cooler with ice packs. After collection, the fecal samples were further transferred in cooler (+4 °C, in average for 4 h) to UNC laboratory and frozen at −80 °C before used for DNA extraction.
DNA was isolated from feces by modified protocol of the DNA clean-up Kit (Qiagen DNeasy Blood and Tissue extraction kit, Qiagen, Valencia, CA, USA), with mechanical disruption of bacterial cells followed by chemical extraction of DNA as previously described (Carroll et al., 2011). The concentration of isolated DNA was determined by using a Nanodrop spectrophotometer (Thermo Scientific, Asheville, NC, USA) before phylogenetic microarray analysis.
Bacterial phylogenetic microarray analysis
Microbial analysis was performed by using the phylogenetic HITChip microarray (Agilent, Palo Alto, CA, USA), which targets the V1 and V6 hypervariable regions of the 16S ribosomal RNA gene, as described and benchmarked previously (Rajilić-Stojanović et al., 2009; Ringel-Kulka et al., 2013). In brief, the 16S ribosomal RNA gene was amplified from sample DNA, transcribed and labeled, and then fragmented before hybridization on the microarray. The signal intensity was quantified by Agilent Feature Extraction software (version 10.7.3.1) after scanning. Sample-wise normalization and quality control was performed using in-house scripts in R statistical software (Lahti et al., 2014). Samples with high reproducibility (>0.95 Pearson’s correlation coefficient for technical replicates) were accepted for further analysis. Min–max algorithm (Bolstad et al., 2003; Han, 2011) was used to normalize between samples.
The HITChip bacterial phylogenetic microarray allows analysis at several taxonomic levels (Rajilić-Stojanović et al., 2009). In this study, we used the genus level (L2, defined to consist of phylotypes having >90% sequence similarity of the 16S RNA gene) and phylum levels (L1, corresponding to a bacterial phylum or different Clostridium clusters within Firmicutes) to analyze microbial composition. Furthermore, oligo-level data were used to analyze microbial diversity, richness, evenness and stability, as described previously (Hill, 1973; McCune et al., 2002; Satokari et al., 2014). The diversity, evenness and stability were quantified by Shannon’s index (Hill, 1973), Pielou’s evenness index (McCune et al., 2002) and Pearson’s correlation coefficient (using consecutive sampling points), respectively. Richness was quantified as responding oligos (Satokari et al., 2014). The relative abundances of L1 and L2 phylogenetic groups was calculated as described previously (Ringel-Kulka et al., 2013), and compiled in Supplementary Table 2. The diversity, richness, evenness and stability of microbiota profiles are summarized in Supplementary Table 3.
The relative abundance of butyrate-producing bacteria was obtained by summing the relative abundances of 10 HITChip genus-level bacterial groups, which have been reported to produce butyrate: Clostridium (sensu stricto), Faecalibacterium prausnitzii et rel., Subdoligranulum variable et rel., Anaerostipes caccae et rel., Butyrivibrio crossotus et rel., Coprococcus eutactus et rel., Eubacterium hallii et rel., Eubacterium rectale et rel., Roseburia intestinalis et rel. and Eubacterium cylindroides et rel.
The stabilities were assessed by profile similarity, measured by Pearson correlation of the oligo-level profiles between consequent time points for total microbiota or each phylum. The mean and 95% confidence intervals were calculated by one sample t-test (Supplementary Table 4). Phyla with average Pearson correlation coefficient (cor.) >0.8 were considered as highly stable, whereas phyla with cor. between 0.6 and 0.8 were specified as moderate stable, and phyla with cor. <0.6 were considered to have low stability (Table 1 and Supplementary Table 4).
Linear mixed effects model
In model fitting, a fully specified linear mixed effects (LMEs) model (Pinheiro and Bates, 2000) was first constructed using the R package ‘nlme’ (Pinheiro et al., 2015). The model included probiotics, antibiotics, zBMI, and age as fixed effects and individual as random effect with autocorrelation structure of order 1 to take into account the effect of repeated sampling to explain the outcomes (that is, dependent variable): stability, diversity, richness and evenness of total microbial profile and relative abundance of various phylum-level (L1) and genus-level (L2) bacterial groups. The fully specified model was then compared against reduced models where one fixed effect was removed at a time using likelihood ratio test. If the test was not significant (P>0.05), the simpler model was accepted. The procedure was carried out to obtain the final model where no effects could be removed without significantly affecting the modeling accuracy. From the final model, P-values for single effects and interaction terms of the explanatory variables were extracted. A false discovery rate correction of the P-values was carried out by Benjamini–Hochberg method (Benjamini et al., 2001). Significant effects are summarized in Supplementary Table 5.
Categorized age groups
The significant findings from LME modeling on relative abundance of bacterial groups were confirmed by categorizing the 83 samples from 28 children into four categories according to the age of the child at sampling: 14–24 months (1–2 years (Y); 10 samples), 25–36 months (2–3 Y; 19 samples), 37–48 months (3–4 Y; 16 samples) and 49–59 (4–5 Y; 6 samples). In order to avoid the possible confounding effect of repeated sampling from the same individual in the same age category, only the first sample for each child in each category was included in the analysis. The samples used in the analysis of age categories are specified in Supplementary Table 1.
Core microbiota estimation
The shared microbiota among children (so-called phylogenetic common core; see Salonen et al., 2012) was analyzed using the categorized age group data. The oldest age group was omitted from core microbiota analysis because of small sample size (six samples). The composition of core microbiota was determined as the set of genus-level bacterial groups that are shared by at least 70% of the subjects and have a relative abundance of at least 0.1%. A bootstrap method for estimating core microbiota was applied (Salonen et al., 2012) using 5000 bootstrap samples. The core microbiota (genus-level bacterial groups), the mean abundance and frequency (proportion of bootstrap samples where the group was selected to the core) in each age category are compiled in Supplementary Table 6. In each age category (three age categories for children and one for adults), the inter-individual similarity was calculated by Pearson correlation between samples.
Statistical analysis
Statistical analysis was carried out in R version 2.15.3 by using the packages ‘stats’ and ‘vegan’ (Oksanen et al., 2015). For the comparison between children and adults, the division to age groups was used for assessing total microbiota diversity/richness/evenness, core microbiota, relative abundance of phylum-level/genus-level groups, and inter-individual similarities. The comparison between different age categories and adults was carried out by Kruskal–Wallis and Wilcoxon rank-sum tests (Siegel and Jr, 1988). For all analyses, false discovery rate corrected P-values below 0.05 were considered significant.
Results
Temporal effect of synbiotic intervention and antibiotics on the phylum-level bacterial groups
Overall, age was found to be the major factor affecting the microbiota profiles in children (discussed in detail later). The other factors did not have a drastic effect on the overall microbiota profiles, but had an effect on the relative abundance or stability of three phylum-level bacterial groups. Specifically, the synbiotic supplementation was found to have a minor effect by increasing the stability of Actinobacteria as compared with the placebo group. There was no difference in the overall abundance of the genus Bifidobacterium between the intervention groups, although the probes targeting the administered Bifidobacterium species showed increased levels after the intervention period in the synbiotic group (data not shown). Antibiotics were found to decrease the relative abundance of Clostridium cluster XIVa in the next sample taken after the course (Supplementary Figure 2a, LME P=0.02). In the following samples after the cessation of antibiotic use, the abundance difference is not significant when compared with the baseline or the next sample after antibiotic course (Supplementary Figure 2b). In addition, the stability of Clostridium cluster I was observed to decline with antibiotic use.
No significant increase in the diversity of intestinal microbiota from 1 to 5 years of age
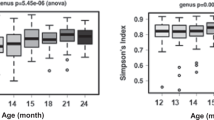
To provide a general view of the temporal changes in intestinal microbiota, we first evaluated microbiota diversity, richness and evenness that all showed high intra-individual variation (Figure 1). To focus on the temporal development, LME was used to estimate the effect of age on the diversity while controlling for the effect of repeated sampling. Overall, no change in microbiota diversity was observed in children from 14 to 59 months of age (corresponding to approximately 1–5 years) (Figure 1a). Moreover, the microbiota diversity was significantly (P<0.01) lower in all age categories in children than in adults (Figure 1). Altogether, it seems that microbiota diversity, including richness and evenness, undergo fluctuations during 1–5 years of age, but there is no developmental trend toward adult-level diversity. As a technical point, it should be noted that the diversity indices are calculated from the HITChip oligo-level data and therefore cannot be directly compared with those based on 16S ribosomal RNA gene sequencing.

The effect of age on (a) diversity, (b) richness and (c) evenness of microbiota. No significant change is observed for diversity, richness or evenness during 1–5 years. Left panel: solid line indicates repeated sampling from the same individual. Dashed line indicates regression fit with LME line (P=NS). Right panel: diversity, richness and evenness plotted in age categories. Samples 1.5 times more than the upper quartile (third quartile) or less than the lower quartile (first quartile) are denoted by circles. Microbial community is significantly less diverse (panel a) and rich (b) in all age categories in children than in adults, whereas being less even (c) in the ‘1–2 Y’ and ‘4–5 Y’ age categories than in adults. Significances were estimated by Wilcoxon rank-sum test, with Benjamini–Hochberg P-value correction.
Major intestinal phyla show discordant development toward an adult-type profile
Although the diversity of microbiota seemed to be at a child-type, presumably transient stage, a number of significant changes in both the abundance and stability of different taxa within the bacterial community were found. First, the stability of the bacterial community was found to increase from 1 to 5 years, since the total microbiota profile similarity between consecutive time points increased with age (P=0.03 in LME, Figure 2). Second, we observed that major intestinal phyla are developing toward adult-type abundancies discordantly (Table 1). For example, Actinobacteria (Figure 3a), Bacilli and Clostridium clusters I and IV were all characterized by child-like abundance levels. However, Clostridium cluster IV showed low stability, whereas Clostridium cluster I, Actinobacteria and Bacilli had high stability (Table 1, Figures 4 and 5). Within Actinobacteria, the largest genus Bifidobacterium had a significantly (P=8.4E–05) higher abundance in children aged 1–2 Y than in adults and its abundance did not change significantly from 1 to 5 years (Supplementary Figure 3). On the other hand, the abundances of Clostridium cluster XIVa (Supplementary Figure 4f) and Proteobacteria (Supplementary Figure 4g) seem to have reached adult-type levels already during childhood (Table 1, Supplementary Figure 4). However, although the stability of Clostridium cluster XIVa was still low (mean cor. =0.50; Figures 4 and 5h), the stability of Proteobacteria was moderate (mean cor.=0.71; Figure 5i). Furthermore, Bacteroidetes (Supplementary Figure 4b), Clostridium cluster III (Figure 3b) and Clostridium cluster XI (Supplementary Figure 4e) seemed to be developing toward adult-type profiles, as denoted by significantly increasing stability or abundance (Table 1). The increasing abundance of Clostridium cluster III was not reflected in a significant abundance increase in any of the individual genus-level groups within the cluster (data not shown). Finally, Verrucomicrobia, represented by a single genus Akkermansia, seemed to be established in abundance already in childhood (Supplementary Figure 5).

Stability of total microbiota in children of 1 to 5 years old. Stability is assessed as the similarity (Pearson correlation) of microbiota profiles between consecutive time points. Each point plotted represents the older age in the pair compared. The time interval between the first plotted time point and the baseline sample point is 4 months. The microbiota stability is increasing significantly with age (LME: P=0.03, slope=0.006 per month). The model fit is indicated by the dashed line.

Relative abundance of Actinobacteria and Clostridium cluster III in different age categories. Between 1 and 5 years, the relative abundances of Actinobacteria (a) do not change (LME: P=NS), whereas the relative abundance of Clostridium cluster III (b) increases significantly (LME: P=0.03). The relative abundances of Actinobacteria (a) in all age categories in children are significantly higher than that of adults, whereas significant differences of Clostridium cluster III (b) between children and adults are observed in the first two age categories: ‘1–2 Y’ (P=0.0008) and ‘2–3 Y’ (P=0.003). Samples 1.5 times more than the upper quartile (third quartile) or less than the lower quartile (first quartile) are denoted by circles.

The stability of phylum-level groups in 1- to 5-year old children. The boxplot shows the distribution of profile similarities (Pearson correlation of oligo-level profiles between consecutive time points) of genus-like taxa with each phylum. The groups with average similarity <0.6 (indicated with the lower horizontal dashed line) are considered as low stability groups, whereas groups with average similarity >0.8 are considered as highly stable (indicated by the upper horizontal dashed line). The groups with similarities between 0.6 and 0.8 are considered moderately stable. Samples 1.5 times more than the upper quartile (third quartile) or less than the lower quartile (first quartile) are denoted by circles.

The development of stability of phylum-level groups in 1- to 5-year-old children. The stability is assessed by similarity of profiles (Pearson correlation) between consecutive time points from oligo-level data. Each plotted point represents the older age in the pair compared. The time interval from the first plotted time point to the baseline sample point is 4 months. Samples from the same individual are connected by solid lines. The development of stability is estimated by LME, and indicated by the dashed line. The stability of Bacteroidetes increases significantly (c, LME: P=0.04), whereas Clostridium cluster XI shows increasing trend (g, LME: P=0.08) and the stabilities of other phylum-level groups remain at similar levels at 1 to 5 years of age (a, b, d, e, f, h and i, LME: P=NS).
Interestingly, among the different phyla, only the Bacteroidetes showed a significant increase in stability from 1 to 5 years (P=0.04; Figure 5c). Thus, Bacteroidetes seemed to be the main component in increasing stability of total microbiota profiles. Within Bacteroidetes, Bacteroides spp. (Supplementary Figure 6a) contributes to the increasing stability of the phylum, whereas Prevotella spp. profiles had consistent, overall high stability in these children (Supplementary Figure 6b).
In addition, the similarity of total microbiota profiles between individuals was assessed by Pearson correlation for each age category, with both qualitative and quantitative estimation. Highest inter-individual similarity was observed for children in the 3–4Y group, differing significantly from the other age groups of children or adults (Supplementary Figure 7).
Core microbiota starts to shape early in life
To estimate the common core of microbiota in children and to assess its compositional development toward an adult-type core, we subdivided and analyzed children in three age categories and compared them with adults. The core microbiota estimation revealed 18 genus-level (L2) groups for the 1–2 Y children that belong to four phylum-level bacterial groups (Figure 6; Supplementary Table 6). Interestingly, 5 out of the 18 genus-level groups were found to be butyrate-producing bacteria. The number of bacterial groups shared between individuals was found to increase with age, and the common core microbiota of children in 2–3 Y and 3–4 Y categories consisted of 23 and 25 genus-level bacterial groups, respectively. After childhood, Bifidobacterium, Streptoccus mitis et rel. and Streptococcus bovis et rel. were replaced by Oscillospira guillermondii et rel., Butyrivibrio crossotus et rel. and Clostridium stercorarium et rel. in the common core of adults (Figure 6). Collectively, the bacterial groups of the core microbiota constituted >70% of the total microbiota, and the summarized abundance was similar in different age categories (Supplementary Figure 8). The results indicate that core microbiota starts to shape early in life, but its assembly is likely to undergo changes after childhood.

The development of core microbiota#. The asterisk (*) indicates typical infant taxa that are diminished in the adult core. The taxa of butyrate-producing bacteria are underlined. # For the core microbiota estimation, see text for details.
Butyrate-producing bacteria increase gradually during childhood
Ten genus-like bacterial groups in the HITChip are known to produce butyrate. To get insight into the temporal development of this important functional group, we summed their relative abundances together. Remarkably, the relative abundance of butyrate-producing bacteria was increasing with age in children (P=0.02 in LME), and their abundance reached similar level to adults before 5 years of age (Supplementary Figure 9). Moreover, the number of butyrate-producing bacterial groups was gradually increasing in the core microbiota (Figure 6), testifying for the diversification and stable establishment of this key functional group in healthy children.
Discussion
The human intestinal microbiota is considered to be an important factor in determining human health. In toddlers, both compositional and functional development of intestinal microbiota are still not well described. In this study, we characterized the fecal microbiota composition of 28 unrelated healthy children (around 1–5 Y) by repeated sampling during a period of up to 1 year. The wide range of sampling ages expanded our observation from subject-specific patterns to general microbiota development trends in children 1–5 years of age.
The children in this study participated also in a synbiotic intervention, which was found to have only a minor effect on fecal microbiota—the only significant effect was that the higher stability of Actinobacteria in the intervention group. The finding is in agreement with several probiotic intervention studies, where limited changes in overall structure of fecal microbiota were observed both in children (Larsen et al., 2011; Nylund et al., 2013) and in adults (Gerritsen et al., 2011; Kim et al., 2013; Lahti et al., 2013). The increased stability of Actinobacteria in the treatment group may have been caused by inulin, which is known to support the growth of bifidobacteria, the main group within the phylum, in the intestine (Veereman, 2007; Salazar et al., 2014).
The use of antibiotics was found to reduce the relative abundance of Clostridium cluster XIVa and the stability of Clostridium cluster I. The result is in line with previous studies showing that antibiotics reduce microbiota stability and diversity (Rehman et al., 2012; Panda et al., 2014). We found that the abundance of Clostridium cluster XIVa was significantly decreased in the next sample after antibiotic course. There is indication that after cessation of antibiotics Clostridium cluster XIVa is recovering, as its abundance is intermediate between the baseline sample before antibiotics and the next sample after the antibiotic course (Supplementary Figure 2b). Thus, the effect of single antibiotic course seems to be temporal and that at least partial recovery takes place during the following weeks and months. Previously, repeated antibiotic courses have been shown to result in an incomplete recovery of microbiota (Dethlefsen and Relman, 2011). In subsequent analysis, the effects of the synbiotic intervention and antibiotics were taken into account as covariates when assessing the temporal development of microbiota.
The main outcome of this study is that intestinal microbiota is not fully established in children up to 5 years of age. First, the bacterial diversity is significantly lower as compared with adults and it does not increase significantly from 1 to 5 years of age. Previously it has been shown that species (operational taxonomic unit) richness sharply increases during the first year of life, especially after weaning (Palmer et al., 2007; Nylund et al., 2013; Yatsunenko et al., 2012). A recent large-scale study showed that, from 1–3 years of age, children from both Amazonas of Venezuela and rural Malawi had an increasingly diverse microbiota, whereas children from the United States in this age category did not show a similar trend (Yatsunenko et al., 2012; Schloss et al., 2014). Our study expands these findings by studying a time course and shows that children from the United States still harbor a significantly less diverse and less stable microbiota as compared with adults up to 5 years of age. Therefore, it could be hypothesized that the western lifestyle may affect the diversification of intestinal microbiota. A number of factors, including overly hygienic conditions, diet and the use of antibiotics, are known to impact microbiota development in infants (Rautava et al., 2012; Nylund et al., 2014). We presume that the children in this study develop a more diverse microbiota later in life; however, at the same time we cannot exclude the possibility that their microbiota does not reach the same level of diversity as in earlier generations.
We found that although the bacterial diversity was at a similar level from 1 to 5 years of age, the microbiota was undergoing gradual transformations. For example, the overall microbial profile stability was increasing, and specific phyla, such as Clostridium clusters III, IV and XI, showed development toward adult-level abundances. However, we also observed that Actinobacteria and Bacilli were stably more abundant in children as compared with adults. Our results contrast the previous perception that intestinal microbiota has reached adult-type configuration by 3 years of age (Yatsunenko et al., 2012). Recently, Bäckhed et al. (2015) studied the development of microbiota in Swedish children from birth to 1 year of age and found that the cessation of BF induces a major diversification of the microbiota and that the microbiota composition at 1 year of age resembles that of adults. However, some bacterial groups still showed differences between 1-year-old children and their mothers (Bäckhed et al., 2015), which is in line with the results of our study, showing that microbiota maturation continues after 1–2 years of age. On the other hand, we found Clostridium cluster XIVa and Proteobacteria to be at adult-like level already in 1–2 years old children. Thus, we conclude that major intestinal phyla seem to develop toward an adult-type abundance and configuration discordantly, and the microbiota maturation exceeds 5 years of age.
Actinobacteria, especially bifidobacteria, are typically found in high quantities in infants (Roger et al., 2010; Arrieta et al., 2014; Bergström et al., 2014). A recent large Danish cohort study showed that the relative abundance of Bifidobacterium spp. decreased significantly from 9 months to 3 years of age. However, the levels were not compared with those in adults (Bergström et al., 2014). BF is known to be one of the major factors affecting microbiota composition in infants and supports the bifidobacterial population in particular (Nylund et al., 2014). In our cohort, the time interval between the cessation of BF and the baseline sampling was on average 21 months. The low number of formula-fed children (n=4) did not allow reliable comparisons, but we did not detect significant differences in the abundances of any genus-level bacterial groups, including bifidobacteria, between BF and formula-fed children in baseline samples (data not shown). However, bifidobacteria remained at a significantly higher level in children up to 5 years of age as compared with adults (Supplementary Figure 3). The effect of the synbiotic intervention in the study cohort is only minor and does not explain the elevated levels, as similar effect was detected in both intervention groups—even during the intervention the bifidobacteria levels were not different between the groups. The observed elevated bifidobacterial levels in early life are potentially beneficial as previous reports suggest a protective role of bifidobacteria against excessive weight gain and neuropsychiatric disorders in later life (Kalliomäki et al., 2008; Pärtty et al., 2015).
The overall stability of microbiota was found to increase over the different age groups over time, that is, from 1 to 5 years of age. However, the microbiota similarities, both in short and long term, were still remarkably lower than previously observed for western adults by using the same microarray technique for microbiota assessment (Jalanka-Tuovinen et al., 2011; Rajilić-Stojanović et al., 2013; Fuentes et al., 2014; Satokari et al., 2014). Furthermore, we found that the most predominant groups within Firmicutes, Clostridium clusters XIVa and IV, had temporally instable profiles, whereas other major phyla, Actinobacteria, Bacteroidetes, Proteobacteria, appeared more stable in children. In adults, all these groups with the exception of Proteobacteria have been described to be relatively stable (Faith et al., 2013; Rajilić-Stojanović et al., 2013). The low stability of Clostridium cluster XIVa and IV in children implies that the species assemblies and taxa abundances within these phyla are still undergoing rearrangements, and thus have not reached a mature, adult-like status. Interestingly, among all phyla, Bacteroidetes was the only one that showed increasing stability, and thus this phylum and more specifically Bacteroides spp. seem to be the main contributors to stability increase of total microbiota profiles. The increasing stability of Bacteroidetes (that is, Bacteroides spp.) may indicate that it is developing toward established, individual-specific species assembly with high stability, as revealed for adults (Faith et al., 2013; Rajilić-Stojanović et al., 2013). Further, the highest inter-individual similarity in 3–4 years children (Supplementary Figure 7) may result from a more established microbiota as compared with younger children, and longer consumption of daycare center meals. Previously, it has been shown that the gut microbiota of elderly people alter shortly after institutionalization, demonstrating the impact of diet and environment (Claesson et al., 2012). Taken together, our results show that the maturing intestinal ecosystem of children is characterized by discordant temporal development of bacterial phyla toward adult-type profiles.
It has been suggested that, despite the highly individual-specific microbiota profiles, people share a common subset of relatively highly abundant bacterial species/groups, the so-called core microbiota (Salonen et al., 2012). In this study, a common core comprising four phylum-level groups (Actinobacteria, Bacilli, Clostridium clusters IV and XIVa) was detected in all child age categories and adults, which indicates the emergence of core microbiota already at around 1 year of age. The common core in children increased with age from 18 to 25 genus-level taxa and developed toward adult-like composition. Similar to our results, Vallès et al. (2014) recently reported the expansion of core microbiota during childhood. However, Vallès et al. (2014) reported a much larger core, 49 genera in 13 1-year-old children, which is most likely due to a different definition of the core as compared with our study. In the Vallès et al. study, the core includes genera that are present in all individuals without any abundance threshold, whereas we applied a threshold of >0.1% of relative abundance and 70% prevalence of the genus-like groups. We found that Bifidobacterium spp., S. mitis et rel. and S. bovis et rel. were replaced by O. guillermondiii et rel., B. crossotus et rel. and Cl. stercorarium et rel. in the adult core. The first mentioned three bacterial groups are typical infant taxa and present in adults in much lower quantities, which explains their disappearance from the adult core (Roger and McCartney, 2010; Arrieta et al., 2014; Bergström et al., 2014).
The abundance of butyrate-producing bacteria was found to increase from 1 to 5 Y, implying the establishment of an important functional group. Butyrate serves as an energy source for host intestinal epithelium cells and upregulates the expression of tight junction proteins (Ulluwishewa et al., 2011; Leonel and Alvarez-Leite, 2012; Plöger et al., 2012), thereby fortifying epithelial barrier function. Furthermore, butyrate promotes the differentiation and proliferation of regulatory T cells, and inhibits nuclear factor-kB activation (Leonel and Alvarez-Leite, 2012; Plöger et al., 2012; Louis et al., 2014), that is, act as anti-inflammatory agent. Although butyrate is mostly described to have positive effects, in an experimental model the improvement of necrotizing enterocolitis was associated with the decrease of butyrate (Butel et al., 2002). On the other hand, in preterm infants the overall number of fecal volatile organic compounds was found to be reduced in subjects who developed NEC (Garner et al., 2009).
In summary, our study shows that the human intestinal microbiota is not established before 5 years of age in US children: the diversity, core microbiota and different bacterial phyla are still developing toward adult-type configuration. The gradual development of microbiota may reflect the stepwise development of host physiology. For example, the most important cellular components of the gastrointestinal immune system are already present in fetal life (Tourneur and Chassin, 2013), but a newborn still has immature intestinal mucosal layer, immunological and metabolic functions (Fernandez et al., 2011; Jakobsson et al., 2014; Levy and Wynn, 2014; Pichichero, 2014). After a few months, infants are able to absorb fat-soluble compounds efficiently because of maturation of bile salt (Fernandez et al., 2011). Furthermore, the full maturation and activation of immune system requires the stimulation of microbiota during many years after birth. For example, the activity of natural killer cells normalizes at 4–5 years of age and IgG and IgA reach adult-like serum levels only at the age of 5–6 years and 10 years, respectively (Jarva and Meri, 2011). The remarkable maturation process of the gastrointestinal physiology and immunology is likely to affect the microbiota; and vice versa, microbiota is likely to be involved in these maturation events (Maynard et al., 2012). On the other hand, nutritional requirements keep rather similar from 1 to 6 years of age (ASPEN Board of Directors and the Clinical Guidelines Task Force, 2002) and relatively stable dietary patterns may serve as a stabilizing factor for the microbiota. In our study, the synbiotic administration was found to have minor effects on the fecal microbiota and because of the intervention trial and occasional antibiotics use, the study population may not have been ideal to investigate microbiota development in controlled environment. On the other hand, the administration of synbiotics and/or antibiotics is not rare among western children, and thus the study represents real-life conditions. Overall, we conclude that the maturation of gut microbiota exceeds 5 years of age. It can be presumed that profound changes in the host physiology during adolescence will further shape the microbiota toward an individual-specific, mature configuration.
References
Arrieta MC, Stiemsma LT, Amenyogbe N, Brown EM, Finlay B . (2014). The intestinal microbiome in early life: health and disease. Front Immunol 5: 427.
ASPEN Board of Directors and the Clinical Guidelines Task Force. (2002). Guidelines for the use of parenteral and enteral nutrition in adult and pediatric patients. J Parenter Enteral Nutr 26 (1 Suppl): 1SA–138SA.
Azad MB, Bridgman SL, Becker AB, Kozyrskyj AL . (2014). Infant antibiotic exposure and the development of childhood overweight and central adiposity. Int J Obes 38: 1290–1298.
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I . (2001). Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125: 279–284.
Bergström A, Skov TH, Bahl MI, Roager HM, Christensen LB, Ejlerskov KT et al. (2014). Establishment of intestinal microbiota during early life: a longitudinal, explorative study of a large cohort of Danish infants. Appl Environ Microbiol 80: 2889–2900.
Bolstad BM, Irizarry RA, Astrand M, Speed TP . (2003). A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinforma Oxf Engl 19: 185–193.
Butel MJ, Waligora-Dupriet AJ, Szylit O . (2002). Oligofructose and experimental model of neonatal necrotizing enterocolitis. Br J Nutr 87: S213e9.
Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P et al. (2015). Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17: 690–703.
Carroll IM, Ringel-Kulka T, Keku TO, Chang YH, Packey CD, Sartor RB et al. (2011). Molecular analysis of the luminal- and mucosal-associated intestinal microbiota in diarrhea-predominant irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol 301: G799–G807.
Cheng J, Kalliomäki M, Heilig HGHJ, Palva A, Lähteenoja H, de Vos WM et al. (2013). Duodenal microbiota composition and mucosal homeostasis in pediatric celiac disease. BMC Gastroenterol 13: 113.
Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E et al. (2011). Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA 108 (Suppl 1): 4586–4591.
Claesson MJ, Jeffery IB, Conde S, Power SE, O’Connor EM, Cusack S et al. (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature 488: 178–184.
Cucchiara S, Iebba V, Conte MP, Schippa S . (2009). The microbiota in inflammatory bowel disease in different age groups. Dig Dis Basel Switz 27: 252–258.
Dethlefsen L, Relman DA . (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic pertubation. Proc Natl Acad Sci USA 108 (Suppl 1): 4554–4561.
Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N et al. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci USA 107: 11971–11975.
Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL et al. (2013). The long-term stability of the human gut microbiota. Science 341: 1237439.
Fallani M, Amarri S, Uusijarvi A, Adam R, Khanna S, Aguilera M et al. (2011). Determinants of the human infant intestinal microbiota after the introduction of first complementary foods in infant samples from five European centres. Microbiol Read Engl 157: 1385–1392.
Fernandez E, Perez R, Hernandez A, Tejada P, Arteta M, Ramos JT . (2011). Factors and mechanisms for pharmacokinetic differences between pediatric population and adults. Pharmaceutics 3: 53–72.
Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H et al. (2011). Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis 17: 179–184.
Fuentes S, van Nood E, Tims S, Heikamp-de Jong I, ter Braak CJF, Keller JJ et al. (2014). Reset of a critically disturbed microbial ecosystem: faecal transplant in recurrent Clostridium difficile infection. ISME J 8: 1621–1633.
Garner CE, Wer AK, Elasouad K, Power F, Greenwood R, Ratcliffe NM . (2009). Analysis of faecal volatile organic compounds in preterm infants who develop necrotizing enterocolitis: a pilot study. J Pediatr Gastroenterol Nutr 49: 559e65.
Gerritsen J, Smidt H, Rijkers GT, de Vos WM . (2011). Intestinal microbiota in human health and disease: the impact of probiotics. Genes Nutr 6: 209–240.
Grönlund MM, Gueimonde M, Laitinen K, Kociubinski G, Grönroos T, Salminen S et al. (2007). Maternal breast-milk and intestinal bifidobacteria guide the compositional development of the Bifidobacterium microbiota in infants at risk of allergic disease. Clin Exp Allergy 37: 1764–1772.
Han J . (2011) Data Mining: Concepts and Techniques. Morgan Kaufmann Press: Burlington.
Hill MO . (1973). Diversity and evenness: a unifying notation and its consequences. Ecology 54: 427–432.
Jakobsson HE, Rodríguez-Piñeiro AM, Schütte A, Ermund A, Boysen P, Bemark M et al. (2014). The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep 16: 164–177.
Jalanka-Tuovinen J, Salonen A, Nikkilä J, Immonen O, Kekkonen R, Lahti L et al. (2011). Intestinal microbiota in healthy adults: temporal analysis reveals individual and common core and relation to intestinal symptoms. PLoS One 6: e23035.
Jarva H, Meri S . (2011) Immune defense of different ages (Finnish) In: Hedman K (ed) Immunologia. Duodecim Press: Helsinkipp 187–195.
Kalliomäki M, Collado MC, Salminen S, Isolauri E . (2008). Early differences in fecal microbiota composition in children may predict overweight. Am J Clin Nutr 87: 534–538.
Kim SW, Suda W, Kim S, Oshima K, Fukuda S, Ohno H et al. (2013). Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing. DNA Res Int J Rapid Publ Rep Genes Genomes 20: 241–253.
Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R et al. (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA 108 (Suppl 1): 4578–4585.
Ladirat SE, Schols HA, Nauta A, Schoterman MHC, Keijser BJF, Montijn RC et al. (2013). High-throughput analysis of the impact of antibiotics on the human intestinal microbiota composition. J Microbiol Methods 92: 387–397.
Lahti L, Salojärvi J, Salonen A, Scheffer M, de Vos WM . (2014). Tipping elements in the human intestinal ecosystem. Nat Commun 5: 4344.
Lahti L, Salonen A, Kekkonen RA, Salojärvi J, Jalanka-Tuovinen J, Palva A et al. (2013). Associations between the human intestinal microbiota, Lactobacillus rhamnosus GG and serum lipids indicated by integrated analysis of high-throughput profiling data. PeerJ 1: e32.
Larsen N, Vogensen FK, Gøbel R, Michaelsen KF, Abu Al-Soud W, Sørensen SJ et al. (2011). Predominant genera of fecal microbiota in children with atopic dermatitis are not altered by intake of probiotic bacteria Lactobacillus acidophilus NCFM and Bifidobacterium animalis subsp. lactis Bi-07. FEMS Microbiol Ecol 75: 482–496.
Leonel AJ, Alvarez-Leite JI . (2012). Butyrate: implications for intestinal function. Curr Opin Clin Nutr Metab Care 15: 474–479.
Levy O, Wynn JL . (2014). A prime time for trained immunity: innate immune memory in newborns and infants. Neonatology 105: 136–141.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI . (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444: 1022–1023.
Louis P, Hold GL, Flint HJ . (2014). The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12: 661–672.
Matamoros S, Gras-Leguen C, Le Vacon F, Potel G, de La Cochetiere MF . (2013). Development of intestinal microbiota in infants and its impact on health. Trends Microbiol 21: 167–173.
Maynard CL, Elson CO, Hatton RD, Weaver CT . (2012). Reciprocal interactions of the intestinal microbiota and immune system. Nature 489: 231–241.
McCune B, Grace JB, Urban DL . (2002) Analysis of Ecological Communities. Mjm Software Design Press: Gleneden Beach, OR.
Nylund L, Nermes M, Isolauri E, Salminen S, de Vos WM, Satokari R . (2015). Severity of atopic disease inversely correlates with intestinal microbiota diversity and butyrate-producing bacteria. Allergy 70: 241–244.
Nylund L, Satokari R, Nikkilä J, Rajilić-Stojanović M, Kalliomäki M, Isolauri E et al. (2013). Microarray analysis reveals marked intestinal microbiota aberrancy in infants having eczema compared to healthy children in at-risk for atopic disease. BMC Microbiol 13: 12.
Nylund L, Satokari R, Salminen S, de Vos WM . (2014). Intestinal microbiota during early life—impact on health and disease. Proc Nutr Soc 73: 457–469.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB et al. (2015), vegan: Community Ecology Package.
Olivares M, Neef A, Castillejo G, Palma GD, Varea V, Capilla A et al. (2014). The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut 64: 406–417.
Palmer C, Bik EM, DiGiilio DB, Relman DA, Brown PO . (2007). Development of the human infant intestinal microbiota. PLoS Biol 5: e177.
Panda S, El khader I, Casellas F, López Vivancos J, García Cors M, Santiago A et al. (2014). Short-term effect of antibiotics on human gut microbiota. PLoS One 9: e95476.
Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I et al. (2006). Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118: 511–521.
Pichichero ME . (2014). Challenges in vaccination of neonates, infants and young children. Vaccine 32: 3886–3894.
Pinheiro J, Bates D, DebRoy S, Sarkar D, R Core Team. (2015), nlme: Linear and Nonlinear Mixed Effects Models. R package version 3.1-120 http://CRAN.R-project.org/package=nlme.
Pinheiro J, Bates D . (2000) Mixed-Effects Models in S and S-PLUS. Springer Press: New York.
Plöger S, Stumpff F, Penner GB, Schulzke JD, Gäbel G, Martens H et al. (2012). Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann NY Acad Sci 1258: 52–59.
Pärtty A, Kalliomäki M, Wacklin P, Salminen S, Isolauri E . (2015). A possible link between early probiotic intervention and the risk of neuropsychiatric disorders later in childhood—a randomized trial. Pediatr Res 77: 823–828.
Rajilić-Stojanović M, Heilig HGHJ, Molenaar D, Kajander K, Surakka A, Smidt H et al. (2009). Development and application of the human intestinal tract chip, a phylogenetic microarray: analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ Microbiol 11: 1736–1751.
Rajilić-Stojanović M, Heilig HGHJ, Tims S, Zoetendal EG, de Vos WM . (2013). Long-term monitoring of the human intestinal microbiota composition. Environ Microbiol 15: 1146–1159.
Rautava S, Luoto R, Salminen S, Isolauri E . (2012). Microbial contact during pregnancy, intestinal colonization and human disease. Nat Rev Gastroenterol Hepatol 9: 565–576.
Rehman A, Heinsen FA, Koenen ME, Venema K, Knecht H, Hellmig S et al. (2012). Effects of probiotics and antibiotics on the intestinal homeostasis in a computer controlled model of the large intestine. BMC Microbiol 12: 47.
Ringel-Kulka T, Cheng J, Ringel Y, Salojärvi J, Carroll I, Palva A et al. (2013). Intestinal microbiota in healthy U.S. young children and adults—a high throughput microarray analysis. PLoS One 8: e64315.
Ringel-Kulka T, Kotch JB, Jensen ET, Savage E, Weber DJ . (2015). Randomized, double-blind, placebo-controlled study of synbiotic yogurt effect on the health of children. J Pediatr 166: 1475–1481.
Roger LC, Costabile A, Holland DT, Hoyles L, McCartney AL . (2010). Examination of faecal Bifidobacterium populations in breast- and formula-fed infants during the first 18 months of life. Microbiol Read Engl 156: 3329–3341.
Roger LC, McCartney AL . (2010). Longitudinal investigation of the faecal microbiota of healthy full-term infants using fluorescence in situ hybridization and denaturing gradient gel electrophoresis. Microbiol Read Engl 156: 3317–3328.
Salazar N, Dewulf EM, Neyrinck AM, Bindels LB, Cani PD, Mahillon J et al. (2014). Inulin-type fructans modulate intestinal Bifidobacterium species populations and decrease fecal short-chain fatty acids in obese women. Clin Nutr 34: 501–507.
Salonen A, Salojärvi J, Lahti L, de Vos WM . (2012). The adult intestinal core microbiota is determined by analysis depth and health status. Clin Microbiol Infect Off Publ Eur Soc Clin Microbiol Infect Dis 18 (Suppl 4): 16–20.
Satokari R, Fuentes S, Mattila E, Jalanka J, de Vos WM, Arkkila P . (2014). Fecal transplantation treatment of antibiotic-induced, noninfectious colitis and long-term microbiota follow-up. Case Rep Med 2014: 913867.
Schloss PD, Iverson KD, Petrosino JF, Schloss SJ . (2014). The dynamics of a family’s gut microbiota reveal variations on a theme. Microbiome 2: 25.
Siegel S, Castellan Jr NJ . (1988) Nonparametric Statistics for The Behavioral Sciences. McGraw-Hill Humanities/Social Sciences/Languages Press: New York.
Toh MC, Allen-Vercoe E . (2015). The human gut microbiota with reference to autism spectrum disorder: considering the whole as more than a sum of its parts. Microb Ecol Health Dis 26: 26309.
Tourneur E, Chassin C . (2013). Neonatal immune adaptation of the gut and its role during infections. Clin Dev Immunol 2013: 270301.
Turnbaugh PJ, Gordon JI . (2009). The core gut microbiome, energy balance and obesity. J Physiol 587: 4153–4158.
Ulluwishewa D, Anderson RC, McNabb WC, Moughan PJ, Wells JM, Roy NC . (2011). Regulation of tight junction permeability by intestinal bacteria and dietary components. J Nutr 141: 769–776.
Vallès Y, Artacho A, Pascual-Garcia A, Ferrús ML, Gosalbes MJ, Abellán JJ et al. (2014). Microbial succession in the gut: directional trends of taxonomic and functional change in a birth cohort of Spanish infants. PLoS Genetics 10: e1004406.
Veereman G . (2007). Pediatric applications of inulin and oligofructose. J Nutr 137: 2585S–2589S.
Verdam FJ, Fuentes S, de Jonge C, Zoetendal EG, Erbil R, Greve JW et al. (2013). Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obes Silver Spring Md 21: E607–E615.
Voreades N, Kozil A, Weir TL . (2014). Diet and the development of the human intestinal microbiome. Front Microbiol 5: 494.
Weber TK, Polanco I . (2012). Gastrointestinal microbiota and some children diseases: a review. Gastroenterol Res Pract 2012: 676585.
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M et al. (2012). Human gut microbiome viewed across age and geography. Nature 486: 222–227.
Acknowledgements
The HITChip teams in Wageningen University and in the University of Helsinki are acknowledged for excellent technical assistance and scientific discussions. The financial support from the Academy of Finland (grant no. 138902, 258439 and 283088 for RS) and the Spinoza award of the Netherlands Organization for Scientific Research (WMdV) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Cheng, J., Ringel-Kulka, T., Heikamp-de Jong, I. et al. Discordant temporal development of bacterial phyla and the emergence of core in the fecal microbiota of young children. ISME J 10, 1002–1014 (2016). https://doi.org/10.1038/ismej.2015.177
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.177
This article is cited by
-
Physiological benefits of Akkermansia muciniphila under high-altitude hypoxia
Applied Microbiology and Biotechnology (2023)
-
Socioeconomic disparities and household crowding in association with the fecal microbiome of school-age children
npj Biofilms and Microbiomes (2022)
-
Gut microbiota as the key controllers of “healthy” aging of elderly people
Immunity & Ageing (2021)
-
Gut microbiome, liver immunology, and liver diseases
Cellular & Molecular Immunology (2021)
-
Supplementation of Lactobacillus early in life alters attention bias to threat in piglets
Scientific Reports (2021)
