
Abstract
In this work, we study the diversity of bathypelagic microbial eukaryotes (0.8–20 μm) in the global ocean. Seawater samples from 3000 to 4000 m depth from 27 stations in the Atlantic, Pacific and Indian Oceans were analyzed by pyrosequencing the V4 region of the 18S ribosomal DNA. The relative abundance of the most abundant operational taxonomic units agreed with the results of a parallel metagenomic analysis, suggesting limited PCR biases in the tag approach. Although rarefaction curves for single stations were seldom saturated, the global analysis of all sequences together suggested an adequate recovery of bathypelagic diversity. Community composition presented a large variability among samples, which was poorly explained by linear geographic distance. In fact, the similarity between communities was better explained by water mass composition (26% of the variability) and the ratio in cell abundance between prokaryotes and microbial eukaryotes (21%). Deep diversity appeared dominated by four taxonomic groups (Collodaria, Chrysophytes, Basidiomycota and MALV-II) appearing in different proportions in each sample. Novel diversity amounted to 1% of the pyrotags and was lower than expected. Our study represents an essential step in the investigation of bathypelagic microbial eukaryotes, indicating dominating taxonomic groups and suggesting idiosyncratic assemblages in distinct oceanic regions.
Similar content being viewed by others


Introduction
The bathypelagic region of the deep ocean, defined as the water column between 1000 and 4000 m, is the largest habitat on Earth, but remains relatively unexplored (Arístegui et al., 2009). Despite its huge volume, it is a relatively homogeneous environment in terms of environmental conditions (Angel, 1993). Thus, pressure for a given depth is constant, as well as temperature (range of −1 to 3 °C), salinity (34.3 to 35.1) and dissolved oxygen concentration (2.4 to 5.7 mg l−1). The deep ocean holds many and varied life forms, mostly linked to microbial food webs (Azam et al., 1983; Michaels and Silver, 1988; Tanaka et al., 2005), dependent on resources like particulate and dissolved organic matter and inorganic nutrients. These resources vary at broad regional scales across the bathypelagic zone depending on the flux of organic compounds from the surface, the age of the water mass and the occasional presence of benthic inputs, such as those from hydrothermal vents (Hansell et al., 2009; Hamme and Emerson, 2013), conforming the niche structure supporting microbial life (Nagata et al., 2010).
Heterotrophic microbial eukaryotes, generally considered as bacterial grazers, are the most important component of bathypelagic microbial communities after prokaryotes. In a recent study, we estimated the average cell abundance (14±1 cells ml−1) and biomass (50±14 pg C ml−1) of microbial eukaryotes in bathypelagic samples (Pernice et al., 2015). These deep ocean assemblages were numerically dominated by cells between 2 and 5 μm in size. It is worth to remember that these cells counts may be underestimated given that a percentage of cells may not survive the pressure changes during recovery (Edgcomb et al., 2011a). Yet, the diversity of these bathypelagic assemblages remains still poorly resolved.
Relatively few papers have analyzed the diversity of bathypelagic microbial eukaryotes using clone libraries of 18S ribosomal RNA (rRNA) genes and Sanger sequencing, an approach that is inherently limited in the amount of sequences generated. Most of these studies focused on the water column, always in a regional scale, either by using universal eukaryotic primers (López-García et al., 2001; Stoeck et al., 2003; Countway et al., 2007; Not et al., 2007; Edgcomb et al., 2011b) or group-specific primers (Bass et al., 2007; Lara et al., 2009). Other diversity studies were done in benthic environments, such as deep-sea subsurface and surface sediments (Edgcomb et al., 2011a; Salani et al., 2012) or in hydrothermal vents (Edgcomb et al., 2002; Sauvadet et al., 2010). The bathypelagic microbial eukaryotic diversity has also been studied by fluorescence in situ hybridization staining (Morgan-Smith et al., 2011; Morgan-Smith et al., 2013). Although fluorescence in situ hybridization provides useful quantitative information of given taxa, it remains restricted to the taxa targeted by the available probes, which depend on previous sequencing information, and therefore never targets the complete assemblage. High-throughput sequencing now allows a much more exhaustive assessment of microbial diversity. The Roche 454-pyrosequencing technique (Margulies et al., 2005) has been used in studies targeting the 18S rRNA gene of microbial eukaryotes inhabiting marine epipelagic waters (Edgcomb et al., 2011b; Logares et al., 2012; Kilias et al., 2013; Massana et al., 2015) and freshwaters (Charvet et al., 2012). This approach includes a PCR step, known to be prone to biases like DNA polymerase errors or primer selectivity, that may affect final amplicon ratios (Acinas et al., 2005). Yet, metagenomics now allows a PCR-free approach to investigate microbial diversity, which is based on extracting 18S ribosomal DNA (rDNA) reads (miTags when using Illumina) from metagenomes (Logares et al., 2014). Combining tag-sequencing and miTags provides a more robust view of microbial diversity, as tag-sequencing can recover low abundance taxa, whereas miTags provides a less biased representation of most abundant taxa.
Here we use pyrosequencing to investigate deep-sea microbial eukaryotic assemblages from the 0.8 to 20 μm size fraction originating from 27 stations located in the Atlantic, Indian and Pacific oceans. Samples were taken during the Malaspina-2010 Circumnavigation Expedition (Pernice et al., 2015). Our study is the first attempt to describe exhaustively the global diversity of bathypelagic heterotrophic microbial eukaryotes by high-throughput sequencing.
Materials and methods
Global sampling of bathypelagic microbial eukaryotes
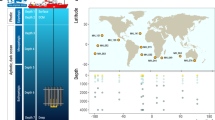
Samples of bathypelagic heterotrophic microbial eukaryotes were collected in 27 stations visited during the Malaspina-2010 Circumnavigation Expedition (Figure 1), conducted from December 2010 to July 2011 on board R/V Hespérides (Duarte, 2015). Bathypelagic seawater was collected with Niskin bottles attached to a rosette sampling system fitted with a Seabird 0911Plus CTD probe, including temperature, salinity and oxygen sensors. Fourteen samples were taken at 4000 m, 11 at depths between 3000 and 4000 m and 2 were shallower: 2400 m in station 62 and 2150 m at station 82 because of bottom depth constrains. Seawater was prefiltered through a 200 μm mesh placed at the end of the hose of the Niskin, and through a 20 μm mesh in a funnel. Between 100 and 120 l of the 20 μm filtrate was then filtered with a peristaltic pump on 142 mm Millipore (Billerica, MA, USA) polycarbonate filters of 0.8 μm pore-size. Filters were flash frozen in liquid nitrogen and stored at −80 °C until processed in the lab.

Map of the stations sampled during the Malaspina-2010 cruise that are used for diversity analysis of bathypelagic microbial eukaryotes. Each station is colored according to the dominant taxonomic groups in the sample: one color if a single group represents >75% of the pyrotags, two (or three) colors if the sum of the groups is >50%. Groups with >20% are always displayed. The top half circle indicates the group at higher abundance, followed by the lower half circle and the inner circle.
We sampled three principal bathypelagic water masses along the track of the Malaspina-2010 Expedition: North Atlantic deep water (NADW), Weddel Sea deep water (WSDW) and Circumpolar deep water (CDW). The proportion of these three water masses in each sample was inferred from its temperature, salinity and oxygen concentration (Catalá et al., 2015). Samples were then clustered in a dendrogram using the inferred deep-water proportions with Pvclust (Suzuki and Shimodaira, 2006), to identify the main water mass types sampled during the cruise. Six types were identified, NADW pure, NADW enriched, CDW pure, CDW enriched, CDW–WSD and WSDW enriched.
DNA extraction, amplification of the 18S rDNA and pyrosequencing
Filters were cut into small pieces and soaked into 3 ml of lysis buffer (40 mM EDTA, 50 mM Tris-HCl, 0.75 M sucrose) at the beginning of the DNA extraction. Afterward, enzymatic digestions were performed. Filters were incubated with Lysozyme (1 mg ml−1 final concentration) at 37 °C for 45 min while slightly shaken, and then Proteinase K (0.2 mg ml−1 final concentration) and sodium dodecyl sulfate (1% final concentration) were added and tubes were incubated at 55 °C for 60 min while slightly shaken. The resulting lysate underwent two steps of standard phenol–chloroform extraction to remove lipids and proteins. After the last centrifugation step, the aqueous phase was collected, concentrated in an Amicon Ultra unit (Millipore) and washed three times with 2 ml sterile deionized water. After the third wash, between 100 and 250 μl of purified total genomic DNA extract was recovered and quantified using a Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA). Typical yields were between 0.08 and 0.58 μg DNA, corresponding to ~1.8 ng l–1 of seawater.
A two-step PCR was required to obtain enough amplicons for pyrosequencing considering the low density of cells in the bathypelagic ocean. We added 2 ng of genomic DNA to triplicate PCR tubes containing dNTPs (0.2 mM), the eukaryotic primers TAReukFWD1 (5′-CCAGCASCYGCGGTAATTCC-3′) and TAReukREV3 (5′-ACTTTCGTTCTTGATYRA-3′) (Stoeck et al., 2010) at 0.5 mM, as well as PCR buffer (1x) in a final volume of 20 μl. Amplicon size was around 380 bp (and variable among taxonomic groups). The first PCR consisted of an initial denaturation step of 98 °C for 2.5 min and then 10 cycles of 45 s at 98 °C, 35 s at 53 °C, 35 s at 72 °C and a final elongation of 10 min at 72 °C. Triplicate amplicons were pooled and purified using the NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel, Hoerdt, France) to a final volume of 18 μl. Then, we did a second PCR step of 20 cycles (45 s at 98 °C, 35 s at 48 °C, 30 s at 72 °C; with a final step of 10 min at 72 °C) using 2 μl of the previous PCR product as template. This second PCR included primers that were the same as before except that they had the 454-specific adaptors, and the forward primer also carried the barcode. We tested with an agarose gel that this second PCR worked correctly, and after this test we run the second PCR step for five replicates, each with 2 μl of template, therefore using most of the first PCR product. Final PCR products were purified, eluted in 30 μl of sterile deionized water and quantified with the Qubit 1.0 fluorometer (Invitrogen, Merelbeke, Belgium). About 200 ng of PCR product were sequenced on a 454 GS FLX+ system (Lifesequencing S.L., Valencia, Spain; http://www.lifesequencing.com). Amplicon sequences produced in this study are publicly available at the European Nucleotide Archive (http://www.ebi.ac.uk/ena/data/view/PRJEB9943).
Processing 454 sequences (pyrotags)
Quality control and demultiplexing of 454 data was processed with QIIME (Caporaso et al., 2010). Owing to the version of the 454-sequencer (GS FLX+), it was not possible to run a denoising algorithm (that is, DeNoiser or AmplicoNoise), so we increased the stringency in the cleaning process. Clean sequences had between 150 and 600 bp in size, no more than 2 mismatches in the primer and no homopolymers longer than 8 bp. Phred quality was analyzed by running windows of 50 bp, and pyrotags were truncated to the last window that had an average Phred >25. Truncated pyrotags shorter than 150 bp were removed. An operational taxonomic unit (OTU) table was constructed by clustering high-quality pyrotags at a 97% similarity threshold using UCLUST v1.22q (Edgar, 2010). The most abundant sequence within each OTU was selected as the OTU representative sequence. The table was manually curated by removing OTUs represented by short sequences (<250 bp) and OTUs that occurred in only one sample, regardless of their abundance (Supplementary Figure S1). OTUs were then taxonomically assigned using three reference databases: SILVA 108 (Quast et al., 2013), PR2 (Guillou et al., 2013) and MAS9013, an in-house database (Pernice et al., 2013; available on request). SILVA 108 was used to exclude prokaryotic 16S rDNA OTUs, whereas PR2 and MAS9013 were used to classify eukaryotic OTUs mostly to the class level. OTUs were assigned to a given group when its representative sequence had a BLAST hit with an e-value below 10−100 with a reference sequence. Above this e-value, OTUs were classified as uncertain. Chimera check was done with UCHIME (Edgar et al., 2011) with default parameters using the MAS9013 database as reference, and the results were carefully evaluated to avoid removing novel OTUs. Thus, OTUs identified as chimeras were kept if their representative sequence had an e-value of 0 against the GenBank database, the similarity between parents was above 90% and the OTU occurred in at least 14 stations or was represented by at least 100 pyrotags.
Community analyses were performed with the R packages Vegan (Okasanen et al., 2013; analyses: non-metric multidimensional scaling (NMDS), Permanova, Shannon index, star plot) and GMT (Wessel et al., 2013; Geodistance test). The number of pyrotags per sample varied from 6625 to 29 926, therefore comparative analyses were done using a second OTU table, subsampled at 6625 pyrotags per sample with rrarefy in Vegan. A Mantel test, based on Bray–Curtis distances and using a Pearson correlation coefficient, indicated that this subsampling set represented very well the original pool of sequences (R=0.98, P<0.0001, Supplementary Figure S2). Repeated NMDS plots based on Bray–Curtis distances were done to keep the plot displaying the minimal stress (Clarke and Ainsworth, 1993).
Analysis of 18S rDNA from metagenomes: miTags
The same genomic DNA extracts were used to obtain metagenomes (except for stations 74, 88 and 109 because of technical reasons) by shotgun sequencing using a Illumina HiSeq2000 (San Diego, CA, USA) platform (2 × 100-bp sequencing) installed at the Department of Energy Joint Genome Institute (JGI) (Walnut Creek, CA, USA). Metagenomic reads including the 18S rDNA (miTags) were extracted as indicated in Logares et al. (2014). Only miTags longer than 100 bp were kept (a total of 19 434 miTags). These miTags were used as queries for a BLAST search against a data set composed by 454 sequences from the same samples. Following this taxonomic identification, miTags were clustered in supergroups (Alveolata, Rhizaria, Stramenopiles, Fungi, Excavata, Amoebozoa) plus Uncertain and Metazoa, and a cluster of other groups (mainly Apusozoa); and then compared with a similar analysis in pyrotags. In a second and more sensitive step, we blasted the 20 most abundant 454 OTUs against the miTag pool to assess their relative abundance. Comparisons were done by averaging the relative contributions in the same set of 24 samples. Metagenomic sequences are available on the Integrated Microbial Genome website (Gs0053074, http://img.jgi.doe.gov/) and a script for the extraction of miTags is available on request.
Results
The diversity of bathypelagic microbial eukaryotes (size fraction of 0.8–20 μm) analyzed by pyrosequencing 18S rRNA genes gave an initial OTU table of 405 270 pyrotags clustered at 97% similarity in 7202 OTUs (Supplementary Figure S1). This value was reduced to 362 910 pyrotags after eliminating OTUs represented by short sequences (<250 bp) and OTUs occurring in only one sample. A posterior chimera check further reduced the number of OTUs by about 10% and that of pyrotags by about 1%. The final OTU table (2481 OTUs and 359 140 pyrotags) was then used to analyze the community diversity in—and between—stations (alpha and beta diversity) and the taxonomic affiliation of bathypelagic microbial eukaryotes. The complete set of 27 samples was considered for the taxonomic analysis, whereas the shallower samples collected at stations 62 and 82 were excluded from the comparative community analysis, thereby based on 25 samples.
Alpha diversity of bathypelagic microbial eukaryote assemblages
The richness observed oscillated between 400 and 1000 OTUs per sample, and rarefaction curves relating the OTUs detected depending on the sequencing effort showed that most samples did not show a sign of saturation (excepting perhaps samples from stations 23 and 131, Figure 2a). The Shannon index, providing an estimate of alpha diversity in each sample, ranged from 0.90 to 4.84 with a mean (±s.e.) value of 3.63±0.17 (Supplementary Figure S3). Shannon indices did not differ across basins, with 3.62±0.45 in the Atlantic, 3.56±0.17 in the Indian and 3.68±0.18 in the Pacific. Samples with the highest Shannon values (above 4.5) were sampled in the subtropical Atlantic (stations 10, 17, 19 and 43), a region that also included two of the samples with lowest diversity (stations 23 and 131). The different water masses also did not show distinct Shannon indices (details not shown). We also performed a rarefaction curve for pooled reads from all samples to produce a view of global diversity, considering the deep ocean as single environment (Figure 2b). The curve showed a clear sign of saturation, with a maximal richness estimated around 2500 OTUs, thereby providing a first-order conservative estimate of the global abundance of bathypelagic microbial eukaryotes OTUs.

Alpha diversity of bathypelagic microbial eukaryotic assemblages as inferred by the analysis of pyrosequences clustered at 97% similarity. (a) Rarefaction curves for each sample, relating the number of OTUs detected as a function of the sequencing effort. (b) Global rarefaction curve relating the OTUs detected to the number of reads from all samples.
Beta diversity of bathypelagic microbial eukaryotic assemblages
To explore microbial eukaryotic community composition among the 25 deepest samples, we performed a NMDS analysis (Figure 3a), based on Bray–Curtis distances calculated from the subsampled OTU table of 6625 pyrotags per sample. Several NMDS plots were done and the one displaying the minimal stress (0.17) was kept. In an attempt to interpret the NMDS clustering, we performed a Permanova test with one continuous variable (distance of the sampling depth to the seafloor) plus three categorized variables (abundance ratio of prokaryotes to microbial eukaryotes, water mass and oceanic region) to identify the contribution of these factors to community structuring. The order of parameters matters so we followed a subordination criterion (ratio <water mass <bottom distance <ocean). We excluded station 10 from this test because we did not have microbial abundance data for this sample. The abundance ratio explained 21% of the variability with high significance (P=0.0001), whereas water mass explained 26% with lower significance (P=0.0018). The distance to bottom and the ocean region explained 6% each, with a significance of about 0.03. Thus, the variance explained by this set of variables was 59%.

Beta diversity in bathypelagic microbial eukaryotic assemblages. (a) NMDS plot based in Bray–Curtis distances displaying community similarity. Samples are grouped according to the water mass: NADW pure (black), NADW enriched (red), CDW pure (yellow), CDW enriched (violet), CDW–WSDW (light blue), WSDW enriched (green). (b) Plot showing Bray–Curtis dissimilarities and geographic distances between assemblages. Average values (±s.e.) in intervals of 1000 km are shown in black.
The NMDS analysis showed a tendency of geographically closer stations to be more clustered in the NMDS plot. To better analyze the relationship between geographic distance and community composition, we performed a Mantel test between the Bray–Curtis dissimilarity matrix and the matrix of geographic distances (Figure 3b). The results showed a weak (R=0.32) but significant (P=0.001) correlation between both variables. In fact, geographically distant samples were almost always different, whereas closer samples were only seldom similar, displaying in some cases high differences (see also Figures 1 and 4).

Relative abundance of the 10 most abundant taxonomic groups in all deep samples. Stations are grouped by their respective water mass.
Taxonomic identification of bathypelagic microbial eukaryotes
Taxonomic analysis was performed at two geographical scales (local and global). To better display the differences in taxonomic composition among samples, we selected the 10 most abundant groups and displayed their relative pyrotag contribution in samples grouped by water mass types (Figure 4). The most evident feature is that fungal tags appeared to be abundant in Pacific and Indian bathypelagic waters (see also Figure 1). Taking together, Ascomycota and Basidiomycota represented on average 25% of pyrotags in CDW enriched samples and 26% in CDW–WSDW samples (with a maximal value of 70% in station 88), whereas they were less important in CDW pure samples (9%) and scarce in the pool of Atlantic samples (<5% in each water mass). Polycystinea (Collodaria and Spumellaria together) were dominant in stations belonging to different water masses, being better represented in NADW enriched (50%), WSDW enriched (46%) and in CDW pure (37%). Chrysophytes preferred NADW pure (47% of pyrotags, with a peak of 82% in station 131) and NADW enriched samples (11%). They were also contributors in the northern part of CDW samples (30% in stations 109-121). MALV-II were widespread in all water types, exhibiting a more constrained relative abundance than the previous groups (between 10% and 18%). The remaining groups (Amoebozoa, Dinophyceae, RAD-B and MALV-I) showed sporadic peaks in few selected stations.
Once analyzed the regional pattern, we decided to average these results in order to give a global view of the bathypelagic ocean. The analysis of pyrotags at the supergroup level indicated that the bathypelagic ocean was dominated by Rhizaria (35%) and Alveolata (25%), followed by Fungi (15%) and Stramenopiles (13%) (Figure 5a). Pyrotags included in the uncertain category represented 1% of the total. The analysis of OTU (clustered at 97% of similarity) was quite different (Figure 5b): Alveolata represented 53% of the OTUs, tripling the number of Rhizaria OTUs (18%), whereas Stramenopiles and Fungi had 5% and 3%, respectively. Interestingly, Excavata, with only 1% of pyrotags, accounted for 8% of the OTUs.

Overview of the diversity of deep microbial eukaryotes at the supergroup taxonomic level. (a) Number of pyrotags per supergroup, averaging the relative abundance in each sample. (b) Number of OTUs clustered at 97% of similarity (OTU97) per supergroup.
Analyzing our data at deeper taxonomic level, we identified 46 class-level groups, also including MAST and MALV ribogroups and the uncertain category (Table 1). At the global level, Collodaria (Rhizaria) was the group with the highest representation (96 688 pyrotags in total contributing to 23.1% pyrotags per sample on average), followed by Chrysophytes (Stramenopiles; 12.2%), MALV-II (Alveolata; 13.1%), Basidiomycota (Fungi; 10.9%) and Dinoflagellates (Alveolata; 7.9%). Interestingly four of the five most abundant groups belonged to different Supergroups. In an attempt to show the degree of novelty within the classes, we also calculated the average of the similarity values of the OTUs against reference sequences: the lowest this average the highest the novelty. A parallel analysis was carried out to calculate the same averaged similarity but weighted by tag abundance. In both cases, Collodaria turned out to contain a high level of intragroup novelty, with an averaged similarity of reads toward reference sequences of 91% (Table 1). For the other dominant groups, this value was generally above 97%. In fact, 59% of taxonomically assigned OTUs had a similarity below 97% against reference sequences, highlighting a considerable level of novelty even within these known groups.
We analyzed the 20 most abundant OTUs in detail (Table 2). Surprisingly, seven of them had high similarity (>97%) with cultured organisms and were present in almost all stations. The most abundant was a colonial Collodaria (45,261 pyrotags) that was 90.3% similar to Collophidium ellipsoides, followed by a Basidiomycota (29099 pyrotags) and a Chrysophyceae (23 512 pyrotags). The two fungal OTUs in the list were very similar to terrestrial strains (98.5% to Tilletiopsis minor and 98.7% to Engyodontium album). The first OTU of MALV-II appears in the thirteenth position. Whereas MALV-II was the most represented group in most samples, it did not contain any OTU with a high dominance, indicating a large diversity and high evenness of the group. Some OTUs detected are very similar to cultured species known to have a cyst stage in their life cycle, such as Pedospumella encystans and Platyamoeba contorta, indicating that the cells sampled may have been dormant or non active. There are evidences that this life stage could be also present in marine fungi (Richards et al., 2012).
Novel diversity in the bathypelagic ocean
A total of 128 OTUs were classified as uncertain because they presented e-values above 10−100 in hits against reference data sets (Table 1). We visually checked whether or not these pyrotags aligned with reference sequences (therefore having the V4-18S rDNA signature), which the majority did. For each uncertain OTU, we identified the closest environmental match and the closest cultured match in a BLAST search against nr-NCBI (Supplementary Table S1). Many of these uncertain OTUs could be assigned to established taxonomic categories (most of them within Rhizaria and Alveolate) in base of the closest cultured match hit, forming probably novel lineages within them. The most abundant group was Collodaria with 15 OTUs, followed by Cercozoa with 14 and Dinophyceae with 6. Some OTUs (11) were metazoan and are not discussed further here. A group of 19 OTUs formed a second level of novelty, as the sequence coverage in the BLAST search was below 50% even with the closest environmental match. These were assigned tentatively to the taxonomy of the closest cultured match as Novel-Group name. Finally, a third level of novelty was represented by a group of 27 OTUs with no BLAST hit in GenBank. Of these, only 5 appeared >10 times and the most abundant had only 229 pyrotags. These were named just as Novel, being potentially new undescribed high-rank groups.
Comparing tag sequencing and metagenomes
MiTags from the same samples were used to compare the relative abundance of taxonomic groups inferred by metagenomics (a PCR-independent approach) and pyrosequencing. Of the 19 434 miTags retrieved, only 3981 showed a similarity above 97% in an alignment of at least 100 bp with the 454-sequencing pool. This percentage (20.5%) is equivalent to the coverage (in percentage) of the amplicon size versus the complete 18S rRNA gene (21.3%). In general, the percentage of supergroups was similar by the two approaches (Figure 6a). Rhizaria and Alveolata had a lower representation in miTags than in pyrotags, whereas Excavata had a much larger representation in miTags (10.7% versus 1.5%). The similarity between the two molecular approaches was generally well supported at local level as well (Supplementary Figure S4 and Supplementary Table S2).

Comparison between pyrosequencing and metagenomics approaches. (a) Relative abundance of each supergroup averaging in both cases the relative abundance in the 24 stations in common. (b) Relationship of the relative abundance of miTags versus pyrotags for the 20 most abundant OTUs.
To compare both approaches at a finer phylogenetic scale, we blasted the 20 most abundant OTUs against the pool of miTag sequences using the same similarity criteria (97% in at least 100-bp alignments). Three abundant Chrysophyte OTUs presented a high sequence similarity and many miTags affiliated indifferently to one of the three, so we pooled the data for them. Remarkably, the resulting 18 abundant OTUs recovered a large fraction of miTags. A total of 2581 miTags produced significant hits, representing 64% of the 3981 miTags that covered the V4 region. At the same time, these abundant OTUs comprised 62% of all pyrotags. In particular, OTU_146 belonging to Collodaria (45 261 pyrotags) retrieved 829 miTags, OTU_6539 (Basidiomycota, 29 099 pyrotags) retrieved 323 miTags and the composite of the three chrysophyte OTUs (23 512 pyrotags) retrieved 419 miTags. When expressed as percentage of the respective data sets (normalized per sample), the relative abundance of these 18 OTUs was highly correlated in the two approaches (Figure 6b), with a very high significance (P<0.0001), an R2 of 0.70, a slope of 0.75±0.13 that is not statistically different from 1 (P=0.06), and an intercept of 0.99±0.51 that is not statistically different from 0 (P=0.07).
Discussion
The results presented comprise, to the best of our knowledge, the first global assessment of the diversity of microbial eukaryotes assemblages in the bathypelagic realm. The geographic coverage sampled during the Malaspina-2010 Circumnavigation Expedition provides a global dimension including all except the polar oceans. Considering the important role of microbial eukaryotes in food webs (Massana, 2011; Richards et al., 2012), our main aim was to characterize global diversity patterns as a first step toward understanding the structure of bathypelagic microbial assemblages and the possible trophic roles of the taxa included. Pyrosequencing has proven to be an adequate tool to assess the composition of microbial communities (Charvet et al., 2012; Logares et al., 2012; Kilias et al., 2013). Our pyrosequencing results are well supported in terms of group relative abundance by miTags. Nevertheless, it is worth mentioning that our study provides a view gathered from screening environmental DNA extracts, whereas a different but complementary view could be obtained by surveying RNA extracts (Stoeck et al., 2007; Not et al., 2009; Massana et al., 2015). The permanence of DNA in the extracellular milieu, different life stages (including dormancy) or differential rDNA copy numbers among species, can generate such different views.
Despite the uniformity of some physical parameters, the deep ocean is not completely a homogeneous environment, as it contains varying amounts of organic matter and other chemical compounds (Hansell et al., 2009; Hamme and Emerson, 2013) that may shape community structure. Considered at a class level, only a few groups dominated the deep microbial eukaryote communities. Thus, Collodaria, Basidiomycota, Chrysophyceae, MALV-II and Dinophyceae accounted together for 69.6% of the pyrotags from the global deep ocean (Table 1) and always explained the majority of reads in each individual sample (Figure 1). The relative proportion of these five groups changed markedly between sites, revealing a high level of variability. At the OTU level, we identified only 42 OTUs that were found in all stations, but together they accounted for 50% of the pyrotags. Although particular classes and OTUs were widespread in the deep ocean, a typical global deep ocean community was not identified, because of the large variation in the relative abundances of these dominant groups.
Our descriptor of alpha diversity, the Shannon index, showed a global average of 3.63±0.17 and did not present a regular pattern along the cruise track nor a relation with environmental parameters. This average was lower than that found in surface marine coastal samples (4.77±0.08, CR Giner, personal communication) at a comparable sequencing effort and using the same sequencing method and data analysis, a difference that had statistical significance (P<0.0001). Rarefaction curves for each individual sample did not saturate, suggesting that our pyrosequencing effort was not exhaustively sampling the microbial eukaryotic diversity in each particular site (Figure 2a). However, this effort appeared satisfactory at a global level (Figure 2b), so we could assume that most abundant taxa present in the deep ocean were encompassed by our samples. In particular, our data suggested a total number of OTUs in the global bathypelagic ocean in the order of 2500. Even assuming that this number could be affected by PCR and sampling biases, we consider this as a relatively low number of taxonomic entities for the largest ecosystem in the planet.
Communities showed a large variability along the cruise, as seen by the differential dominance of taxonomic groups in separate stations (Figures 1 and 4) and the distribution of samples in the NMDS (Figure 3a). Geographic distance explained only a small part (10%) of the genetic variability among samples (Figure 3b), and in fact only a few geographically close stations had a very similar community composition (for example, 17 and 19; 112 and 121). Similarity among nearby samples has been found in other studies of marine microbial eukaryotes at spatial scales of thousands of kilometers (Scheckenbach et al., 2010; Salani et al., 2012) and it is likely caused by similar environmental conditions in nearby sites. In our study, the most important factor explaining microbial eukaryotic composition was the water mass type, which explained 26% of the variability observed, and a similar effect has been reported for marine prokaryotes (Agogué et al., 2008; Varela et al., 2008; Galand et al., 2009; Kirchman et al., 2010). If this was due to relevant differences in chemical composition of the water mass, then our data would agree with the general view of microbial biogeography obtained from surface waters (Massana and Logares, 2013), with the environment selecting for specifically adapted microbes. An alternative explanation would be that the particular history of the water mass is determining microbial composition, suggesting geographical restriction to microbial dispersal.
Unexpectedly the abundance of bacteria, which could be seen as the main food source for deep microbial eukaryotes, did not have a direct effect on taxonomic composition (data not shown). However, the situation changed when considering the ratio of prokaryotes to microbial eukaryotes, which explained 21% of the variability in the community structure with high significance. The average bathypelagic prokaryote:micro-eukaryote ratio (5000 prokaryote cells per microbial eukaryotic cell) is very high when compared with surface samples, where it is about 1000–2000 (Sanders et al., 1992; Pernice et al., 2015) suggesting that predator pressure on prokaryotes may be relatively small in the bathypelagic ocean, particularly for the samples with the highest ratio. Despite the abundances of deep prokaryotes and microbial eukaryotes are partially related, it is likely that microbial eukaryotic heterotrophy is also present in the bathypelagic ocean in the form of osmotrophy and parasitism. These alternative lifestyles are generally linked to particular taxonomic classes, such as fungi or MALV clades, yet a more meticulous taxonomic analysis is required to understand the microbial food-web dynamics in the deep ocean.
There are only a handful molecular reports of deep marine microbial eukaryotes produced by Sanger sequencing, and in these studies the assemblages were dominated by Alveolates, mainly MALV-II (López-García et al., 2001; Not et al., 2007) or Rhizarian (Countway et al., 2007; Sauvadet et al., 2010). The most abundant group in our survey was Collodaria, an order of the class Polycystinea (Rhizaria) that includes species with a colonial lifestyle and without silification (Ishitani et al., 2012). In surface waters, Collodaria normally bear photosynthetic endosymbionts and are categorized as organisms with phototrophic behavior (Stoecker et al., 2009), but we do not have information about their lifestyle in the deep, dark ocean where photosynthetic symbionts are not expected. Probably, the presence of colonies could explain the abundance of these tags in our data set. MALV-II were also abundant in our global survey. This ribogroup contains several examples of parasites, such as Amoebophyra spp. infecting dinoflagellates (Coats and Park, 2002), and parasitism probably is the trophic role characteristic of the entire group (Massana, 2011), perhaps including hosts other than dinoflagellates (Small et al., 2012). Interestingly in Atlantic waters, there was a positive correlation (R2=0.96, P=0.0005) between the relative abundances of MALV-II and Metazoan tags, which could indicate metazoans being infected by MALV-II parasites.
Besides the expected finding of rhizaria and MALV-II, our global survey also unveiled Chrysophytes and Fungi as important members of deep microbial eukaryotic assemblages. Chrysophytes, moderately represented in surface marine waters and seldom detected in deep-sea surveys (Massana and Pedrós-Alió, 2008; del Campo and Massana, 2011), was the second most abundant class. These organisms can be facultative or strict phagotrophs, osmotrophs, or phototrophs (Holen and Boraas, 1996). A few cultured isolates (Pedospumella encystans and Spumella danica) explained 79% of chrysophyte sequences. Chrysophytes are good candidates for being important bacterivores in deep waters.
Fungi have been reported to dominate in deep subsurface sediments on the basis of clone libraries (Edgcomb et al., 2011a) and pyrosequencing rRNA surveys (Orsi et al., 2013), and overall deep-sea fungal diversity appears dominated by Basidiomycota and Ascomycota with probable yeast lifestyle (Bass et al., 2007; Richards et al., 2012). As far as we know, our study is the first identifying fungi as a globally important eukaryotic group in deep seawater, with Basidiomycota being particularly abundant. Considering the careful sampling and methodological processing, and the fact that fungal tags followed a comprehensive distribution in the data set, we excluded the possibility of fungal contamination. Finally, our data highlighted that species of Excavata, particularly Diplonemids, can also be important components of bathypelagic microbial eukaryotic assemblages. These include many bacterivorous forms and are typically more abundant in deep waters than at the surface (Lara et al., 2009), representing up to 15% of fluorescence in situ hybridization-counted cells in bathypelagic microbial eukatyotic assemblages (Morgan-Smith et al., 2013). Excavata were poorly represented in the 454 survey, likely due to a putative negative bias against the group by the V4 primers used (Pawlowski et al., 2011), but reached up to 11% of miTags in the parallel metagenomic analysis. It is interesting to highlight that 60% of OTUs having a representative sequence longer than 450 bp belonged to Excavata, and this longer sequence could cause negative bias during PCR amplification. Excavata cells, cryptic to our PCR-based approach, could thus be important phagotrophs in the deep ocean.
Compared with the better-studied surface microbial eukaryotic community (Massana, 2011), the first obvious difference in bathypelagic assemblages was the paucity of photosynthetic groups. Although sequences from Bacillariophyta, Bolidophyta, Dictyochophyta, Prasinophyceae, Prymnesiophyceae, Raphidophyta and Eustigmatophyceae were retrieved in our survey, together they only represented 0.14% of the pyrotags and 2% of the OTUs. The phototrophic signal in bathypelagic waters is likely due to sinking particles (Agusti et al., 2015), although we do not totally dismiss the idea of some species being facultative heterotrophs. The absence of photosynthetic groups explains the low contribution of Stramenopiles in the global deep community, whereas Alveolate and Rhizaria were represented with percentages similar as in surface (Moon-van der Staay et al., 2001; Not et al., 2007; Massana, 2011). Surprisingly, in a world dominated by heterotrophy, the MASTs ribogroups that are important phagotrophs in the surface ocean (Massana et al., 2014) were virtually undetected. For instance, one of the most abundant and widely distributed ribogroup, MAST-4 (Rodríguez-Martínez et al., 2009), was represented in our data set by only 18 pyrotags. The difference in the composition of taxonomic groups between surface and deep waters implies that there are different factors in the flux of energy and carbon through the microbial loop.
The amount of pyrotags of uncertain identification was only 1% (Figure 5). A tentative taxonomic assignation based on the closest environmental match showed that many of the uncertain OTUs affiliated with the most abundant taxonomic groups, that is, Rhizaria and Alveolata. The truly novel sequences formed only 27 OTUs (Supplementary Table S1). We also investigated the novelty within each class, and found that the novelty was very high in Collodaria, whereas two of the other dominant groups, Fungi and Chrysophytes, showed lower levels of novelty as seen by the analysis of their dominant OTUs that were rather close to cultured species. Taking into account the peculiarity of the deep ocean environment and the sampling of unstudied oceanic basins, the percentage of novel species discovered was lower than expected, suggesting that the bathypelagic ocean contains a limited set, about 2500 OTUs, of microbial eukaryotic taxa.
This study provides the first insight into general diversity patterns of microbial eukaryotes in the bathypelagic ocean. Thanks to the breadth of the Malaspina-2010 expedition we collected a large number of pyrotags from samples in separate marine regions that were further validated by a parallel metagenomic survey. We identified a few groups (Collodaria, Chrysophytes, MALV-II and Basidiomycota) dominating microbial eukaryotic communities but mixed in different proportions in each sample. Community differences were partially explained by the water mass they belong to and the abundance ratio between prokaryotes and microbial eukaryotes. As far as we know, the fact that Chrysophytes and fungi dominated some bathypelagic water samples was not reported before. Our research describes the global architecture of bathypelagic microbial eukaryotes communities and provides an essential underpinning to assess the role of eukaryotic microbes in the largest ecosystem on Earth.
References
Acinas S, Sarma-Rupavtarm R, Klepac-Ceraj V, Polz M . (2005). PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl Environ Microbiol 71: 8966.
Agogué H, Brink M, Dinasquet J, Herndl GJ . (2008). Major gradients in putatively nitrifying and non-nitrifying Archaea in the deep North Atlantic. Nature 456: 788–791.
Agusti S, Gonzalez-Gordillo JI, Vaque D, Estrada M, Cerezo MI, Salazar G et al. (2015). Ubiquitous healthy diatoms in the deep sea confirm deep carbon injection by the biological pump. Nat Commun 6: e-pub ahead of print 9 July 2015; doi:10.1038/ncomms8608.
Angel MV . (1993). Biodiversity of the Pelagic ocean. Conserv Biol 7: 760–772.
Arístegui J, Gasol JM, Duarte CM, Herndl GJ . (2009). Microbial oceanography of the dark ocean’s Pelagic realm. Limnol Oceanogr 54: 1501–1529.
Azam F, Fenchel T, Field J, Gray J, Meyer-Reil L, Thingstad F . (1983). The ecological role of water-column microbes in the sea. Mar Ecol Prog Ser 10: 257–263.
Bass D, Howe A, Brown N, Barton H, Demidova M, Michelle H et al. (2007). Yeast forms dominate fungal diversity in the deep oceans. Proc R Soc B 274: 3069–3077.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7: 335–336.
Catalá TS, Reche I, Fuentes-Lema A, Romera-Castillo C, Nieto-Cid M, Ortega-Retuerta E et al. (2015). Turnover time of fluorescent dissolved organic matter in the dark global ocean. Nat Commun 6: 5986.
Charvet S, Vincent WF, Comeau AM, Lovejoy C . (2012). Pyrosequencing analysis of the protist communities in a High Arctic meromictic lake: DNA preservation and change. Front Microbiol 3: 422.
Clarke KR, Ainsworth M . (1993). A method of linking multivariate community structure to environmental variables. Mar Ecol Prog Ser 92: 205–219.
Coats DW, Park MG . (2002). Parasitism of photosynthetic dinoflagellates by three strains of Amoebophyra (Dinophyta): parasite survival, infectivity, generation time and host specificity. J Phycol 38: 520–528.
Countway PD, Gast RJ, Dennet MR, Savai P, Rose JM, Caron DA . (2007). Distinct protistan assemblages characterize the euphotic zone and deep sea (2500 m) of the western North Atlantic (Sargasso Sea and Gulf Stream). Environ Microbiol 9: 1219–1232.
del Campo J, Massana R . (2011). Emerging diversity within chrysophytes, choanoflagellates and bicosoecids based on molecular surveys. Protist 162: 435–448.
Duarte CM . (2015). Seafaring in the 21st century: the Malaspina 2010 Circumnavigation Expedition. Limnol Oceanogr Bull 24: 11–14.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R . (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194–2200.
Edgcomb V, Kysela D, Teske A, de Vera Gomez A, Sogin M . (2002). Benthic eukaryotic diversity in the Guaymas Basin hydrothermal vent environment. Proc Natl Acad Sci USA 99: 7658–7662.
Edgcomb V, Beaudoin D, Gast R, Biddle JF, Teske A . (2011a). Marine subsurface eukaryotes: the fungal majority. Environ Microbiol 13: 172–183.
Edgcomb V, Orsi W, Bunge J, Jeon S, Christen R, Leslin C et al. (2011b). Protistan microbial observatory in the Cariaco basin, Caribbean. I. Pyrosequencing vs Sanger insights into species richness. ISME J 5: 1344–1356.
Galand PE, Casamayor EO, Kirchman DL, Lovejoy C . (2009). Ecology of the rare microbial biosphere of the Arctic Ocean. Proc Natl Acad Sci USA 106 (52): 22427–22432.
Guillou L, Bachar D, Audic S, Bass D, Berney C, Bittner L et al. (2013). The protist ribosomal reference database (PR2): a catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucleic Acids Res 41: 597–604.
Hansell DA, Carlson CA, Repeta DJ, Schlitzer R . (2009). Dissolved organic matter in the ocean: a controversy stimulates new insights. Oceanography 22: 202–211.
Hamme RC, Emerson SR . (2013). Deep-sea nutrient loss inferred from the marine dissolved N2/Ar ratio. Geophys Res Lett 40: 1149–1153.
Holen DA, Boraas ME . (1996) Mixotrophy in chrysophytes. In:Craig D, Sandgren CD, Smol JP, Kristiansen J (eds.) Chrysophyte Algae Ecology, Phylogeny and Development. University Press: Leiden, 1996: pp 119–140.
Ishitani Y, Ujiié Y, de Vargas C, Not F, Takahashi K . (2012). Phylogenetic relationships and evolutionary patterns of the order Collodaria (Radiolaria). PLoS One 7: e35775.
Kilias E, Wolf C, Nöthig E-M, Peeken I, Metfies K . (2013). Protist distribution in the Western Fram Strait in summer 2010 based on 454-pyrosequencing of 18S rDNA. J Phycol 49: 996–1010.
Kirchman DL, Cottrell MT, Lovejoy C . (2010). The structure of bacterial communities in the western Arctic ocean as revealed by pyrosequencing of 16S rRNA genes. Environ Microbiol 12: 1132–1143.
Lara E, Moreira D, Vereshchaka A, López-García P . (2009). Pan-oceanic distribution of new highly diverse clades of deep-sea diplonemids. Environ Microbiol 11: 47–55.
Logares R, Audic S, Santini S, Pernice MC, de Vargas C, Massana R . (2012). Diversity patterns and activity of uncultured marine heterotrophic flagellates unveiled with pyrosequencing. ISME J 6: 1823–1833.
Logares R, Sunagawa S, Salazar G, Cornejo-Castillo FM, Ferrera I, Sarmento H et al. (2014). Metagenomic 16S rDNA Illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environ Microbiol 16: 2659–2671.
López-García P, Rodríguez-Valera F, Pedrós-Alió C, Moreira D . (2001). Unexpected diversity of small eukaryotes in deep-sea Antarctic plankton. Nature 409: 603–607.
Margulies M, Egholm M, Altman W, Attiya S, Bader J, Bemben L et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380.
Massana R, Pedrós-Alió C . (2008). Unveiling new microbial eukaryotes in the surface ocean. Curr Opin Microbiol 11: 213–218.
Massana R . (2011). Eukaryotic picoplankton in surface oceans. Annu Rev Microbiol 65: 1–47.
Massana R, del Campo J, Sieracki ME, Audic S, Logares R . (2014). Exploring the uncultured microeukaryote majority in the oceans: reevaluation of ribogroups within stramenopiles. ISME J 8: 854–866.
Massana R, Logares R . (2013). Eukaryotic versus prokaryotic marine picoplankton ecology. Environ Microbiol 15: 1254–1261.
Massana R, Gobet A, Audic S, Bass D, Bittner L, Boutte C et al. (2015). Marine protist diversity in European coastal waters and sediments as revealed by high-throughput sequencing. Environ Microbiol e-pub ahead of print 28 June 2015; doi:10.1111/1462-2920.12955.
Michaels AF, Silver MW . (1988). Primary production, sinking fluxes and the microbial food web. Deep Sea Res I 35: 473–490.
Moon-van der Staay SY, De Wachter R, Vaulot D . (2001). Oceanic 18S rDNA sequences from picoplankton reveal unsuspected eukaryotic diversity. Nature 409: 607–610.
Morgan-Smith D, Herndl GJ, van Aken HM, Bochdansky AB . (2011). Abundance of eukaryotic microbes in the deep subtropical North Atlantic. Aquat Microb Ecol 65: 103–115.
Morgan-Smith D, Clouse MA, Herndl GJ, Bochdansky AB . (2013). Diversity and distribution of microbial eukaryotes in the deep tropical and subtropical North Atlantic Ocean. Deep Sea Res I 78: 58–69.
Nagata T, Tamburini C, Arístegui J, Baltar F, Bochdansky AB, Fonda-Umani S et al. (2010). Emerging concepts on microbial processes in the bathypelagic ocean – ecology, biogeochemistry, and genomics. Deep Sea Res II 57: 1519–1536.
Not F, Gausling R, Azam F, Heidelberg JF, Worden AZ . (2007). Vertical distribution of picoeukaryotic diversity in the Sargasso Sea. Environ Microbiol 9: 1233–1252.
Not F, del Campo J, Balagué V, de Vargas C, Massana R . (2009). New Insights into the Diversity of Marine Picoeukaryotes. PLoS ONE 4: e7143.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB et al. (2013), Vegan: Community Ecology Package. R package version 2.0-7 http://CRAN.R-project.org/package=vegan.
Orsi W, Biddle JF, Edgcomb V . (2013). Deep sequencing of subseafloor eukaryotic rRNA reveals active fungi across marine subsurface provinces. PLoS One 8: e56335.
Pawlowski J, Christen R, Lecroq B, Bachar D, Shahbazkia HR, Amaral-Zettler L et al. (2011). Eukaryotic richness in the Abyss: insights from pyrotag sequencing. PLoS One 6: e18169.
Pernice MC, Logares R, Guillou L, Massana R . (2013). General patterns of diversity in major marine microeukaryote lineages. PLoS One 8: e57170.
Pernice MC, Forn I, Gomes A, Lara E, Alonso-Saez L, Arrieta JM et al. (2015). Global abundance of planktonic heterotrophic protists in the deep ocean. ISME J 9: 782–792.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41: 590–596.
Sanders RW, Caron DA, Berninger UG . (1992). Relationships between bacteria and heterotrophic nanoplankton in marine and fresh waters: an inter-ecosystem comparison. Mar Ecol Prog Ser 86: 1–14.
Small HJ, Shields JD, Reece KS, Bateman K, Stentiford GD . (2012). Morpholmgical and molecular characterization of hematodinium perezi (Dinophyceae: Syndiniales), a Dinoflagellate parasite of the Harbour Crab, Liocarcinus depurator. J Euk Microbiol 59: 54–66.
Richards TA, Jones MDM, Leonard G, Bass D . (2012). Marine fungi: their ecology and molecular diversity. Annu Rev Mar Sci 4: 495–522.
Rodríguez-Martínez R, Labrenz M, del Campo J, Forn I, Jürgens K, Massana R . (2009). Distribution of the uncultured protist MAST-4 in the Indian Ocean, Drake Passage and Mediterranean Sea assessed by real-time quantitative PCR. Environ Microbiol 11: 397–408.
Salani FS, Arndt H, Hausmann K, Nitsche F, Scheckenbach F . (2012). Analysis of the community structure of abyssal kinetoplastids revealed similar communities at larger spatial scales. ISME J 6: 713–723.
Sauvadet A-L, Gobet A, Guillou L . (2010). Comparative analysis between protist communities from the deep-sea pelagic ecosystem and specific deep hydrothermal habitats. Environ Microbiol 12: 2946–2964.
Scheckenbach F, Hausmann K, Wylezich C, Weitere M, Arndt H . (2010). Large-scale patterns in biodiversity of microbial eukaryotes from the abyssal sea floor. Proc Natl Acad Sci USA 107: 115–120.
Stoeck T, Taylor G, Epstein S . (2003). Novel eukaryotes from a permanently anoxic Cariaco basin (Caribbean Sea). Appl Environ Microbiol 69: 5656–5663.
Stoeck T, Bass D, Nebel M, Christen R, Jones MDM, Breiner H-W et al. (2010). Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol Ecol 19: 21–31.
Stoeck T, Zuendorf A, Breiner H-W, Behnke A . (2007). A Molecular Approach to Identify Active Microbes in Environmental Eukaryote Clone Libraries. Microb Ecol 53: 328–339.
Stoecker D, Johnson M, deVargas C, Not F . (2009). Acquired phototrophy in aquatic protists. Aquat Microb Ecol 57: 279–310.
Suzuki R, Shimodaira H . (2006). Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22: 1540–1542.
Tanaka T, Rassoulzadegan F, Thingstad TF . (2005). Analyzing the trophic link between the mesopelagic microbial loop and zooplankton from observed depth profiles of bacteria and protozoa. Biogeosciences 2: 9–13.
Varela MM, Van Aken HM, Herndl GJ . (2008). Abundance and activity of Chloroflexi-type SAR202 bacterioplankton in the meso- and bathypelagic waters of the (sub)tropical Atlantic. Environ Microbiol 10: 1903–1911.
Wessel P, Smith WHF, Scharroo R, Luis J, Wobbe F . (2013). Generic mapping tools: improved version released. Eos Trans Am Geophys Union 94: 409–410.
Acknowledgements
This project was supported by the Spanish Ministry of Economy and Competitiveness through project Consolider-Ingenio Malaspina-2010 (CSD2008–00077) to CMD and FLAME (CGL2010-16304) to RM. RL was supported by a Juan de la Cierva Fellowship (JCI-2010-06594, Spanish Ministry of Economy and Competitiveness, Spain). Sequencing at the JGI was supported by US Department of Energy (DOE) JGI 2011 Microbes Program grant CSP 602 grant to SGA. The work conducted by the US Department of Energy Joint Genome Institute is supported by the Office of Science of the US Department of Energy under Contract No. DE-AC02-05CH11231. Additional funding was provided by the Spanish Ministry of Economy and Competitivity grant MALASPINOMICS (CTM2011-15461-E). The authors were also supported by the Generalitat de Catalunya Research Group grants 2014SGR/1179. We thank our colleagues Xose A. Álvarez-Salgado, Teresa S. Catalá, Guillem Salazar, Francisco Cornejo-Castillo, Encarna Borrull, Cristina Díez, Elena Lara, Irene Forn, Dolors Vaqué, Eugenio Fraile, Verónica Benitez, the crew and the chief scientists of the various cruises for helpful collaboration.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Pernice, M., Giner, C., Logares, R. et al. Large variability of bathypelagic microbial eukaryotic communities across the world’s oceans. ISME J 10, 945–958 (2016). https://doi.org/10.1038/ismej.2015.170
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.170
This article is cited by
-
Water mass age structures the auxiliary metabolic gene content of free-living and particle-attached deep ocean viral communities
Microbiome (2023)
-
Community dynamics of microbial eukaryotes in intertidal mudflats in the hypertidal Bay of Fundy
ISME Communications (2023)
-
Beyond the limits of the unassigned protist microbiome: inferring large-scale spatio-temporal patterns of Syndiniales marine parasites
ISME Communications (2023)
-
Comparison of Deep-Sea Picoeukaryotic Composition Estimated from the V4 and V9 Regions of 18S rRNA Gene with a Focus on the Hadal Zone of the Mariana Trench
Microbial Ecology (2022)
-
Revealing the full biosphere structure and versatile metabolic functions in the deepest ocean sediment of the Challenger Deep
Genome Biology (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}