
Abstract
To characterize the activity and interactions of methanotrophic archaea (ANME) and Deltaproteobacteria at a methane-seeping mud volcano, we used two complimentary measures of microbial activity: a community-level analysis of the transcription of four genes (16S rRNA, methyl coenzyme M reductase A (mcrA), adenosine-5′-phosphosulfate reductase α-subunit (aprA), dinitrogenase reductase (nifH)), and a single-cell-level analysis of anabolic activity using fluorescence in situ hybridization coupled to nanoscale secondary ion mass spectrometry (FISH-NanoSIMS). Transcript analysis revealed that members of the deltaproteobacterial groups Desulfosarcina/Desulfococcus (DSS) and Desulfobulbaceae (DSB) exhibit increased rRNA expression in incubations with methane, suggestive of ANME-coupled activity. Direct analysis of anabolic activity in DSS cells in consortia with ANME by FISH-NanoSIMS confirmed their dependence on methanotrophy, with no 15NH4+ assimilation detected without methane. In contrast, DSS and DSB cells found physically independent of ANME (i.e., single cells) were anabolically active in incubations both with and without methane. These single cells therefore comprise an active ‘free-living’ population, and are not dependent on methane or ANME activity. We investigated the possibility of N2 fixation by seep Deltaproteobacteria and detected nifH transcripts closely related to those of cultured diazotrophic Deltaproteobacteria. However, nifH expression was methane-dependent. 15N2 incorporation was not observed in single DSS cells, but was detected in single DSB cells. Interestingly, 15N2 incorporation in single DSB cells was methane-dependent, raising the possibility that DSB cells acquired reduced 15N products from diazotrophic ANME while spatially coupled, and then subsequently dissociated. With this combined data set we address several outstanding questions in methane seep microbial ecosystems and highlight the benefit of measuring microbial activity in the context of spatial associations.
Similar content being viewed by others


Introduction
Methane is a potent greenhouse gas, and its consumption by microbes in methane seep sediment reduces its release into the overlying water column (Reeburgh, 2007). The oxidation of methane in seep sediments is mediated primarily by three groups of anaerobic methanotrophic archaea (ANME): ANME-1 (Orphan et al., 2002), ANME-2 (Boetius et al., 2000; Orphan et al., 2001b) and ANME-3 (Niemann et al., 2006; Lösekann et al., 2007). Although they can be detected as single cells or monospecific aggregates (Orphan et al., 2002, Lösekann et al., 2007), ANME, and particularly ANME-2 and ANME-3, are typically found in direct physical association with Deltaproteobacteria. ANME-2 associate with three distinct groups of putatively sulfate-reducing Deltaproteobacteria: (1) SEEP-SRB1, members of Desulfosarcina/Desulfococcus (DSS) within the Desulfobacteraceae (Boetius et al., 2000; Orphan et al., 2001a); (2) SEEP-SRB2, a deeply branching deltaproteobacterial group originally described as the Eel-2 group (Orphan et al., 2001a; Kleindienst et al., 2012); and (3) SEEP-DBB, within the Desulfobulbaceae (DSB) (Pernthaler et al., 2008; Green-Saxena et al., 2014). ANME-3 have been shown to associate with relatives of SEEP-SRB3 within the DSB (Niemann et al., 2006; Lösekann et al., 2007). Although the chemical interaction between ANME-2/-3 and the associated Deltaproteobacteria remains an area of active research (Moran et al., 2008; Milucka et al., 2012), the associated Deltaproteobacteria are traditionally thought to mediate sulfate reduction, consuming the reduced products of ANME-2/-3 methane oxidation and driving the thermodynamic favorability of the anaerobic oxidation of methane (Hoehler et al., 1994; Boetius et al., 2000; Orphan et al., 2001b; Hallam et al., 2004; Alperin and Hoehler, 2009).
Deltaproteobacteria, including DSS and DSB, are also detected as physically independent (i.e., single) cells within methane seep sediment, comprising 5–20% (>108 cells per cm3) of the non-ANME-associated population (Knittel et al., 2003; Schreiber et al., 2010; Kleindienst et al., 2012). Although often referred to as ‘free-living’ cells, it is not known if single DSS and DSB are active when physically independent of ANME. In one study, it was suggested that the presence of abundant single DSB cells at an oil field site was because of disruption of the ANME–DSB association, rather than the presence of a truly free-living population (Schreiber et al., 2010). If they are active, how their physiology compares with their ANME-associated counterparts, and particularly whether they are dependent on methane, as ANME-associated Deltaproteobacteria appear to be (Nauhaus et al., 2002; Dekas et al., 2009, 2014), remain intriguing questions.
Direct characterization of the activity of single versus ANME-associated Deltaproteobacteria is challenging, because standard experiments (e.g., sulfate reduction rates, enzyme activity and phylogenetic or isotope analyses of bulk-extracted biomolecules, including DNA, RNA or lipids) cannot differentiate between phylogenetically similar organisms occupying distinct spatial niches. Most studies investigating single seep Deltaproteobacteria have focused on their abundance, distribution and phylogenetic identity, without assessing their activity or ecological function (Knittel et al., 2003; Lösekann et al., 2007; Schreiber et al., 2010; Kleindienst et al., 2012). There is therefore a great deal of uncertainty related to what fraction of the single-cell assemblage is active, what their metabolic capabilities entail and if and how they interact with the ANME-Deltaproteobacteria consortia.
Recently, nitrogen fixation, the biological conversion of N2 to NH3, has been observed in methane seep sediment from the Eel River Basin (ERB) and Mound 12 Costa Rica (Dekas et al., 2009, 2014). Although ANME-2 archaea were identified as the primary diazotrophs at Mound 12, a wide diversity of dinitrogenase reductase (nifH) gene sequences have been described from Mound 12 as well as other deep-sea methane seeps, raising the possibility that multiple members of the community are able to fix nitrogen (Dang et al., 2009; Dekas et al., 2009, 2014; Miyazaki et al., 2009). Putatively sulfate-reducing Deltaproteobacteria, including DSS and DSB, are candidates for additional seep N2 fixation, both because some seep-recovered nifH sequences show high similarity to those of cultured diazotrophic sulfate-reducing Deltaproteobacteria, and beacause N2 fixation mediated by sulfate-reducing bacteria has been observed in shallow marine sediments (Bertics et al., 2010, 2012; Fulweiler et al., 2013). Therefore, if single seep Deltaproteobacteria are indeed active, they may be a source of bioavailable nitrogen to the seep ecosystem.
Here, we sought to determine if single DSS and DSB cells in methane seep sediment (1) are active, (2) are dependent on methane and/or ANME activity and (3) if they fix nitrogen. To this end, we investigated the activity of bacteria and archaea in sediment collected at Mound 12 Costa Rica in microcosm experiments amended with methane or argon, and either 15NH4+ or 15N2. We investigated the microbial community composition, activity and response to methane with an analysis of DNA and RNA (rRNA and mRNA). We then measured anabolic activity and/or diazotrophic ability in single DSB, single DSS, ANME-2-associated DSS, ANME-1 and ANME-2 in the presence and absence of methane with fluorescence in situ hybridization coupled to secondary ion mass spectrometry (FISH-NanoSIMS). With this combined data set, we were also able to address several additional outstanding questions in seep microbial ecosystems, including if ANME-2 are anabolically active in the absence of methane, whether ANME-1 and ANME-2 display differences in anabolic activity and if there is a phylogenetic diversity of active diazotrophs in Costa Rican seep sediment.
Materials and methods
Sample collection
Seafloor sediment push cores investigated in this study were collected using the manned submersible Alvin and R/V Atlantis in October 2006 (cruise number AT15-11) within methane seep sites in the ERB Southern Ridge (~40°47.192′N, 124°35.706′W; 520 m water depth; 5 °C in situ water temperature) and in January 2010 (cruise number AT15-59) at Mound 12, Costa Rica (~8°55.8′N, 84°18.7′W; 988 m water depth; 5 °C in situ water temperature). Sediment cores were immediately stored at 4 °C and extruded from push core liners in 3 cm increments on-board within 2 h after recovery of the submersible. Sediment samples were either stored in Mylar bags flushed with argon (Ar) at 4 °C (ERB) or immediately combined with Ar-sparged filtered seawater and aliquoted into anaerobic serum bottles (CR). Push cores used in this study are listed in Supplementary Table 1.
15N-labeling microcosm incubations
As described in Dekas et al. (2009, 2014), sediments were homogenized with Ar-sparged artificial marine media (ERB) or with filtered bottom water collected near core sampling (CR). Sediment slurries were aliquoted into 140 ml (ERB) or 35 ml (CR) serum bottles with butyl stoppers, crimped and amended with methane (CH4) or argon (Ar) (to an overpressure of 2 atm), as well as 15N2 (5.2%, ERB, or 2%, CR, of headspace) or 15NH4+ (2 mm, ERB, or 1 mm, CR, final concentration), and then stored at 4 °C in the dark. 15N2 gas was supplied by Cambridge Isotopes (Tewksbury, MA, USA; NLM-363, lot no. I1-10077 or I1-10798). Subsamples of sediment slurry were collected via needle and syringe at at 0, 28, 84 and 168 days (ERB), and at 4, 63, 139 and 275 days (CR). Subsamples for FISH were fixed with 2% paraformaldehyde overnight at 4 °C, washed with phosphate-buffered saline (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4·7H2O, 2 mm KH2PO4) and ethanol, and stored at −20 °C in 100% ethanol. Subsamples for nucleic acid extraction were immediately flash frozen in liquid nitrogen in 2 ml cryovials and stored at −80 °C. Incubation details are summarized in Supplementary Table 1.
A total of 46 incubations were conducted with CR sediment from six push cores. Bulk rates of nitrogen fixation (15N2 incorporation), anabolic activity (15NH4+ incorporation) and sulfide production were measured over time, and previously reported in Dekas et al. (2014). Incubations with the highest rates of nitrogen fixation (CR8 and CR15, both amended with 15N2 and CH4), and incubations from the same sample as CR15 but amended differently (CR17, amended with 15N2 and Ar, and CR18, amended with NH4+ and Ar), were selected for further analysis in the current study. CR45 (amended with 15N2 and Ar) was also analyzed here, to confirm NanoSIMS results observed in CR17. All analyses reported here were conducted on CR incubations, except the FISH-NanoSIMS analysis of ANME-1 cells, which were conducted on ERB incubations, because of low numbers of ANME-1 cells in the CR sediment.
The time points investigated were selected based on trends in 15N incorporation and sulfide production and over time: 139-day subsamples (~5 months, Costa Rica incubations) and 168-day subsamples (6 months, ERB incubations). Given the long doubling time of ANME-Deltaproteobacteria consortia (estimated 3–7 months; Girguis et al., 2005; Nauhaus et al., 2007; Krueger et al., 2008; Orphan et al., 2009), long incubations times are necessary to observe the synthesis of new biomass via 15N incorporation, particularly when N2 serves as nitrogen source (Dekas et al., 2009). Non-target, DAPI (4,6-diamidino-2-phenylindole)-stained cells were analyzed to ensure that excessive recycling of substrates did not occur.
DNA and RNA extractions and reverse transcription reactions
DNA and RNA were extracted simultaneously from 1 ml of frozen sediment slurry using the RNA Powersoil Total RNA Isolation Kit (MOBIO Laboratories, Carlsbad, CA, USA; cat. no. 12866-25) and the RNA Powersoil DNA Elution Accessory Kit (MOBIO Laboratories; cat. no. 12867-25). The extraction was performed according to the manufacturer’s instructions, with the following modification: after the addition of solution SR2, the mixture was divided into four 2-ml screw top tubes and cells mechanically lysed using a Bio 101 FastPrep FP120 bead beater (Thermo Electron Corporation, Milford, MA, USA) for 45 s at a speed of 5.5 three times. The RNA extracts were treated with the Ambion TURBO DNA-free Kit (ThermoFisher Scientific, Waltham, MA, USA; cat. no. AM1907), and cleaned using the Qiagen RNeasy Kit (Hilden, Germany; cat. no. 74104), following the RNA Clean-up Protocol provided by the manufacturer. Reverse transcription of RNA to cDNA was completed using Superscript III First Strand Synthesis Supermix (ThermoFisher Scientific; cat. no. 18080-400).
DNA and cDNA clone libraries
Traditional Sanger sequencing of clone libraries was performed to obtain near full-length 16S rRNA sequences (in contrast to the shorter reads provided by higher throughput alternatives) to enable superior phylogenetic analysis and comparison with regions used in FISH oligonucleotide probe design. Fourteen libraries were generated from RNA and DNA extracted simultaneously from incubations of AD4587 PC6 sediment of the 3–6 cm horizon at the 20-week time point: CR15 (15N2 with CH4 headspace) and CR17 (15N2 with Ar headspace). Twenty-five microliters of PCR reactions containing 1 μl each of 10 μm forward and reverse primer, 1 μl template (5–16 ng DNA or cDNA), 2.5 μl of 10 × ExTaq PCR buffer (Takara, Clontech Laboratories, Inc., Mountain View, CA, USA), 0.3 μl of 5 U μl−1 ExTaq (Takara), 0.5 μl of 10 mm dNTPs (New England Biolabs, Ipswich, MA, USA), 0.5 μl of 10 μg μl−1 bovine serum albumin and 18.2 μl water were performed. The following primer sets were used: nifHf_10aa and nifHr_132aa (Mehta et al., 2003) to target dinitrogenase reductase (nifH), mcrA_F and mcrA_R modified from (Luton et al., 2002) to target methyl coenzyme M reductase A (mcrA), AprA-1-FW and AprA-5-RV (Meyer and Kuever, 2007) to target adenosine-5′-phosphosulfate reductase α-subunit (aprA) and 27 F and 1492 R modified from Lane (1991) to target the 16S rRNA gene. Primer sequences are listed in Supplementary Table 2, and PCR conditions in Supplementary Table 3. No amplicon was visible in any of the RNA-only reactions (no-RT reaction) when 4 μl were run on an agarose gel. PCR products were plate purified (Millipore Multiscreen filter plates; ref. no. MSNU03010), ligated with the Invitrogen TOPO TA Cloning Kit (ThermoFisher Scientific; cat. no. K457501) and transformed using Top Ten chemically competent Escherichia coli cells. Picked colonies were grown overnight in Luria–Bertani broth and amplified using M13 primers for 30 cycles. The M13 products were visualized to confirm the correct size insert, plate purified and sent for unidirectional sequencing using T3 primers at Laragen Sequencing (Culver City, CA, USA). For the 16S rRNA library, clones that were identified as Deltaproteobacteria after unidirectional sequencing were sent for reverse sequencing with T7 primers. All unique, full-length sequences were deposited into GenBank with the following accession numbers: KR813881–KR814285 (16S rRNA), KR020406–KR020496 (nifH), KR812737–KR813018 (aprA) and KR812576–KR812736 (mcrA). Sequences derived from CR15 begin 'CH4–' and sequences derived from CR17 begin 'Ar–'.
Phylogenetic analysis and identification
All sequences were trimmed, examined for quality and stitched (16S rRNA deltaproteobacterial sequences only) using Sequencher software (Gene Codes Corporation, Ann Arbor, MI, USA). Two 16S rRNA gene trees were inferred by maximum likelihood using PhyML package (Guindon et al., 2010) and the HKY evolutionary model in the software program ARB version 5.5 (Ludwig et al., 2004). The bacterial positional variability filter was provided within the SSURef-111-SILVA-NR database (Quast et al., 2013). The reliability of the trees was estimated by bootstrapping in Geneious version 7.0.4 (Kearse et al., 2012) using PhyML maximum likelihood, the HKY model and 100 replicates. The 16S rRNA identification of all clones was also checked using the SSURef-119-SILVA-NR database. The aprA sequences were translated in ARB and aligned with MUSCLE (MUltiple Sequence Comparison by Log-Expectation). The aprA phylogeny was computed using MrBayes (Ronquist et al., 2012). Convergence was determined by an average standard deviation of split frequencies <0.01. The nifH and mcrA sequences were translated in Geneious and aligned using ClustalW. The nifH and mcrA trees were generated by maximum likelihood (PhyML) using the LG substitution model (Le and Gascuel, 2008), with branch support estimated using the approximate likelihood ratio test (Anisimova and Gascuel, 2006).
FISH-NanoSIMS
FISH-NanoSIMS was performed on paraformaldehyde-fixed ANME-Deltaproteobacteria consortia and/or single cells from CR8, CR17, CR18 and CR45 (139-day time point), and ERB1C and ERB5A (168-day time point) using previously described protocols (Dekas and Orphan, 2011). CR8 rather than CR15 was investigated to pair the single-cell FISH-NanoSIMS analyses performed here with the ANME-2 consortia FISH-NanoSIMS analyses performed previously (reported in Dekas et al., 2014). CR8 and CR15 are analogous incubations: both contained sediment collected under microbial mats from Mound 12, both sediment inoculum horizons were proposed to be the methane–sulfate transition zone based on ANME abundance, their sediment inoculum showed similar diversity in nifH sequences and both showed methane-dependent sulfide production and nitrogen fixation (Dekas et al., 2014).
Sediment was centrifuged in a 1:1 phosphate-buffered saline:Percoll gradient (Sigma-Aldrich, St Louis, MO, USA; P4937), followed by filtration on a 0.2-μm polyvinylidene fluoride filter (Durapore Membrane Filter; EMD Millipore, Hayward, CA, USA). Damp filters were flipped onto slides, depositing cells on the slide surface (custom-cut glass squares coated with indium tin oxide or glass rounds). Catalyzed reporter deposition fluorescence in situ hybridization (CARD-FISH) was conducted following the protocols in Pernthaler et al. (2002) and Pernthaler and Pernthaler (2007) using the following horseradish peroxidase-labeled probes: DSS_658 (Boetius et al., 2000), seepDBB_653 (Green-Saxena et al., 2014) and ANME-1_350 (Boetius et al., 2000). Probe sequences and formamide concentrations are listed in Supplementary Table 2. Tyramides conjugated with Alexa Fluor 488 or Alexa Fluor 546 were used in the CARD-FISH amplification reactions. Hybridizations with DSS_658 and seepDBB_653 were conducted sequentially on the same samples. Cells were counterstained with DAPI. Consortia containing both cells positively hybridized with the DSS_658 probe and unhybridized, DAPI-stained cells were identified as ANME-DSS consortia for NanoSIMS analysis. To support this identification, mono-label FISH experiments (as described in Orphan et al., 2002) were conducted on sediment from the same incubations using probes DSS_658 and EelMS_932 (targeting ANME-2) (Boetius et al., 2000). These experiments demonstrated that >90% of cell clusters containing DSS cells were indeed ANME-2-DSS aggregates (Supplementary Figure 1B). Cells of interest were imaged and mapped using 60 × (PlanApo; Olympus, Shinjuku, Tokyo), 40 × (UPlanFLN; Olympus) and 10 × (Plan-Neofluar; Zeiss, Jena, Germany) objectives on a Delta Vision RT microscope and Softworx software (Applied Precision, Issaquah, WA, USA).
A CAMECA NanoSIMS 50 l housed at Caltech (Pasadena, CA, USA), operated with a mass resolving power of ~5000, was used to analyze specific cells identified with FISH. Cells deposited on glass were gold coated before NanoSIMS analysis. A Cs+ primary ion beam (2–8 pA) with a nominal spot size of 100–200 nm was used to rastor over cells of interest. Seven masses were collected: 12C−, 13C−, 14N12C−, 14N13C−, 28Si−, 31P− and 32S− using electron multipliers. Raster images of 9–100 μm2 were collected at 256 × 256 or 512 × 512 pixels resolution, for 0.5–5 h. Clostridia spores with known isotopic composition (previously analyzed by isotope-ratio mass spectrometry) were used as standards. Images were processed using L’Image software (developed by L Nittler, Carnegie Institution of Washington, Washington, DC, USA).
Results and Discussion
Combining DNA and RNA Investigations with FISH-NanoSIMS
Investigating the activity of microbes within natural samples is a focus of environmental microbiology, and a key to understanding the role of microbes in biogeochemical cycles. Here, we investigated the occurrence (DNA) and expression (RNA) of four key genes in methane seep sediment from Mound 12 Costa Rica incubated with either CH4 or Ar to assess the potential activity of seep microorganisms. We analyzed 16S bacterial rRNA, to target the bacterial community, aprA, to target sulfur-cycling microbes, mcrA, to target methanotrophs and methanogens and nifH, to target organisms capable of N2 fixation (diazotrophs).
Although RNA provides a more accurate assessment of microbial activity than DNA, the detection of rRNA and/or mRNA is still an imperfect proxy (see discussion in Blazewicz et al., 2013). Ribosomes can be detected in dormant cells (e.g., Sukenik et al., 2012), and posttranscriptional and posttranslational regulation can lead to decoupling of mRNA expression, protein synthesis and enzyme activity (e.g., Kessler and Leigh, 1999; Liang et al., 1991; Waldbauer et al., 2012; Zhang et al., 1993). Additionally, although RNA in deceased cells is generally thought to degrade quickly, the lifetime of transcripts in the environment—particularly in cold anoxic sediments—is poorly understood, and there is some evidence for survival of RNA post- mortem (Fordyce et al., 2013). It is therefore beneficial to combine community-level RNA analysis with direct measurements of cellular activity when possible.
Consequently, we follow our RNA analysis with direct measurements of microbial anabolic activity using FISH-NanoSIMS. We measure uptake of 15N2 and 15NH4+ to directly detect diazotrophic activity (N2 fixation) and overall anabolic activity, respectively (Krueger et al., 2008; Orphan et al., 2009). In addition to providing a more definitive measure of microbial activity, FISH-NanoSIMS observes activity in the context of spatial associations, allowing the differentiation of activity between phylogenetically similar microbes occupying different spatial niches (e.g., Deltaproteobacteria in association with ANME versus physically independent). Despite the benefits of FISH-NanoSIMS analysis, single-cell isotope analysis remains a time-consuming and expensive procedure, precluding the analysis of more than a small subset of cells within a community. The two methods are therefore complimentary: we use the DNA and RNA analysis to obtain a broad community perspective of the organisms present and potentially active under different experimental conditions, and targeted FISH-NanoSIMS analyses to both validate trends seen in our RNA analysis and test specific hypotheses generated by the RNA analysis.
Methanotroph community composition and activity
We detected all three major groups of ANME at Mound 12 (Figures 1a and b). mcrA groups a–e have been paired with the 16S rRNA identities of the ANME previously: mcrA groups a and b belong to ANME-1, c and d to ANME-2c, e to ANME-2a and f to ANME-3 (Hallam et al., 2003; Lösekann et al., 2007; Meyerdierks et al., 2010; Wang et al., 2014). In the methane incubation, ANME-1-affiliated mcrA sequences were recovered from the DNA-based survey (6% of the library), with a single ANME-1-affiliated clone detected in the cDNA library (1% of the library) (Figures 1a and b). ANME-2- and ANME-3-affiliated mcrA sequences together dominated the DNA library (46% and 49%, respectively), but transcripts affiliated with ANME-3 were not detected (Figures 1a and b).

Relative abundance of genes and transcripts detected in 14 clone libraries generated from DNA and cDNA derived from Mound 12 sediment incubated with CH4 or Ar. In (a and b), Deltaproteobacteria are in color, with DSB-affiliated sequences in orange and Desulfobacteraceae-affiliated sequences in yellow. In (c), green bars highlight phylotypes demonstrating higher relative transcription with CH4. Asterisks indicate significant increases as determined by a one-tailed Z-test of proportions: *95% confidence; **99% confidence; n, number of clones sequenced; nm, not measured; nd, none detected. 1Not-incubated, t=0 sediment (data previously appeared in Dekas et al., 2009); 2SRB, sulfate-reducing bacteria; SOB, sulfide-oxidizing bacteria, as defined by Meyer and Kuever (2007); 3grouping as defined by Raymond et al. (2004); 4grouping as defined by Hallam et al. (2003).
mcrA transcripts were also detected in the Ar incubation (Figures 1a and b). Notably, PCR amplification of mcrA in cDNA from the Ar incubation was significantly less than that from the CH4 incubation, determined by visualization of the PCR product via gel electrophoresis. From this library, 18 mcrA sequences were obtained, all belonging to the ANME-2c-affiliated group c/d, and highly similar to mcrA transcripts recovered from the methane treatment (Figures 1a and b and Supplementary Figure 2). Three primary possibilities may explain the detection of these mcrA transcripts in the absence of methane: ANME methanotrophy fueled by in situ methane production, methanogenesis by ANME or low turnover of mcrA transcripts. Methanogenesis by ANME has been proposed in other sediments based on thermodynamic calculations (Alperin and Hoehler, 2009), as well as environmental observations including the detection of ANME-1 16S rRNA and mcrA transcripts within methanogenic zones of sediment cores (Lloyd et al., 2011), direct natural abundance δ13C measurements of ANME-1 and ANME-2 cells (House et al., 2009) and contemporaneous detection of methane production and oxidation in ANME-dominated sediments (Orcutt et al., 2005, 2008; House et al., 2009). However, as described above, detection of transcripts does not definitively indicate activity. We therefore employed FISH-NanoSIMS to determine if the detection of ANME-2-affiliated mcrA transcripts in the Ar incubation coincided with anabolic activity.
ANME-DSS consortia: anabolically active in the absence of CH4?
To determine if ANME-2-DSS consortia were anabolically active without methane, we measured uptake of 15NH4+ by ANME-2-DSS consortia in the Ar incubation via FISH-NanoSIMS. FISH using oligonucleotide probes EelMS_932 and DSS_658 revealed the occurrence of intact, ribosome-containing ANME-2-DSS consortia in the absence of methane for 9 months, without statistically significant changes in abundance or morphology (CR17 and CR18; Supplementary Figure 1). Previous FISH-NanoSIMS analyses showed no 15N incorporation from 15N2 by ANME-2-DSS aggregates in the absence of methane in incubation CR17 (Dekas et al., 2014), but left open the possibility of non-diazotrophic anabolic activity. Here, we demonstrated that general anabolic activity (15NH4+ uptake) by ANME-2-DSS consortia was also below detection after 14 months of incubation without methane (n=4 consortia, including ~550 cells, calculated assuming average cell volume of 1 μm3, CR18); (Figures 2c and 3a).

NanoSIMS analysis of 15N assimilation from 15N2 (a and b, left axis) or 15NH4+ (c, right axis) in ANME-DSS consortia, ANME–DSB consortia, unidentified DAPI-stained single cells, DSS single cells and DSB single cells, recovered from Costa Rica Mound 12 sediment after incubation with the indicated amendments. ‡Data previously published in Dekas et al. (2014), redisplayed here for comparison. The dashed lines indicate natural abundance atom % 15N.

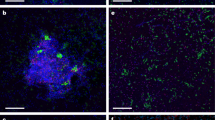
Paired CARD-FISH and NanoSIMS images of methane seep microorganisms incubated with 15NH4+ and Ar (a–c) or 15N2 and CH4 (d–g). a1, b1, c1, d1, e1, f1, g1: CARD-FISH images show DSB (probe seepDBB_653), DSS (probe DSS_658) and ANME-1 (probe ANME-1_350), counterstained with DAPI (blue) as indicated. (a1) An ANME-2-DSS aggregate with probe DSS_658 in red. a2, b2, c2, d3, e2, f2, g2: NanoSIMS images of the same cells show their isotopic (atom % 15N) composition. (d2) A 12C− ion image of the same cells in (d1 and d3), with 12C− counts ranging from 0 to 900 per pixel. The minimum value for all atom % 15N color bars is natural abundance; the maximum varies by image. The isotope images show data only for pixels that exceed a threshold for total 12C− counts (10–30% of the maximum 12C− counts in the image); these areas are enclosed by white outlines and may include more area compared with the cell of interest because of the presence of non-cellular carbon-containing particles and/or extracellular polymeric substance (EPS). In (g2), the arrow and green outline indicate the location of the cell, which was drawn by hand, and determined using the 12C15N− and 32S− ion images (not shown). Scale bars in (a2, d2 and d3) are 5 μm; all others are 1 μm.
The lack of 15NH4+ uptake demonstrates that although they persist without methane, and are detected by FISH, the ANME-2 and associated DSS cells investigated were anabolically dormant without methane. Anabolic dormancy in this subset of the population suggests that ANME-2-mediated methanogenesis, or methanotrophy fueled by in situ methane production, coupled to growth, is not widespread in this community. These results also suggest that ANME-2-associated DSS are typically dependent on ANME activity (and/or directly on methane), and do not fully disassociate or recover the ability to grow independently of ANME/methane even after months in the absence of active methanotrophy. However, because of the targeted nature of FISH-NanoSIMS, even with the analysis of ~550 cells, we cannot eliminate the possibility of 15NH4 incorporation in a subset of the ANME-2- and/or ANME-associated DSS populations not included in our analysis.
Sulfur-cycling bacterial diversity and activity
The bacterial 16S rRNA genes recovered from Mound 12 revealed a bacterial assemblage typical of methane seep habitats, including a large fraction of bacteria likely involved in sulfur cycling (Figures 1a and b). We detected three of the four uncultured, seep-specific putatively sulfate-reducing clades: SEEP-SRB1, SEEP-SRB3 and SEEP-SRB4, all of which expressed rRNA (Figure 4). Interestingly although transcripts belonging to the DSB clades SEEP-SRB3 and Seep-DBB were detected in the cDNA (together comprising up to 18% of the cDNA libraries), no SEEP-SRB3 or Seep-DBB genes were detected in the DNA clone libraries (n=199 clones) (Figures 1a and b). Their presence and ecological contribution may therefore be overlooked by investigations of DNA alone, using conventional cloning and sequencing methods.

16S rRNA gene tree inferred with maximum-likelihood, HKY evolutionary model and 100 bootstraps. Sequences from this study are in bold. cDNA clones are larger and in color, with clones incubated with CH4 in red (incubation CR15), and Ar in blue (incubation CR17). For brevity, only a representative subset of the clones from this study is included in the tree. The total number of clones from each library that fall within the group of the clone shown (defined by the last well-supported branch, and indicated by the black square) is included within parentheses. Purple and green phylogenetic labels indicate DSB and Desulfobacteraceae, respectively. Bootstrap support of 70 or greater is shown. The scale bar indicates the average number of nucleotide substitutions per site. One thousand three hundred and seven nucleotides were used to infer the tree. *A shorter sequence (866–987 bp) that was inserted into the tree by parsimony. **A more detailed version of this portion of the tree can be found in Supplementary Figure 4. The tree was rooted with Aquificaceae species AB026268, GU233444, AP011112, AJ005640 and M83548. NCBI accession numbers are shown.
The aprA gene sequences recovered were split nearly evenly between those of sulfate-reducing bacteria (aprA groups SRB I and II) and sulfide-oxidizing bacteria (aprA groups SOB I and II), but the transcripts were dominated by SRB sequences (Figures 1a and b and Supplementary Figure 3). Sulfate reduction is therefore likely more prevalent than sulfide oxidation in these incubations, consistent with the net production of sulfide observed over time (Dekas et al., 2014). Few DSB aprA sequences were detected in DNA or cDNA, which is surprising given the abundance of DSB 16S rRNA sequences in the cDNA. Although this could suggest that the aprA sequences of the uncultured DSB groups detected do not cluster with the aprA sequences of cultured DSB, the disparity may also be because of mismatches in the aprA primer set to the aprA sequences of seep DSB. The aprA reverse primer contained two or more mismatches to the aprA genes within a DSB-linked metagenomic bin from a Hydrate Ridge methane seep (Connor Skennerton and Victoria Orphan, unpublished data). Therefore, the lack of DSB aprA sequences cannot be commented on.
Differences in bacterial expression with methane: ANME-coupled activity?
Because of the important role Deltaproteobacteria likely play in the anaerobic oxidation of methane, there is great interest in identifying Deltaproteobacteria in metabolic partnerships with ANME. Previous studies have used FISH to visualize bacteria physically associated with ANME (e.g., Boetius et al., 2000; Orphan et al., 2001b; Niemann et al., 2006; Lösekann et al., 2007; Pernthaler et al., 2008; Schreiber et al., 2010; Holler et al., 2011; Kleindienst et al., 2012; Vigneron et al., 2014). In the current study, we utilized a different approach to detect bacterial lineages with ANME-coupled activity, by observing differential 16S rRNA gene expression with and without methane. Bacterial lineages whose metabolisms are positively linked with ANME activity were expected to display higher relative levels of rRNA expression in the presence of methane (i.e., when ANME are active) than without. Unlike FISH, this approach does not require that metabolic partners are physically associated to be detected.
The two bacterial groups with the largest increase in 16S rRNA expression with CH4 were SEEP-SRB1 and SEEP-SRB3 (Figure 1c and Supplementary Figure 4). Indeed, SEEP-SRB1 transcripts were only detected with methane, suggesting that Seep-SRB1 are dependent on methane and/or ANME activity. This is consistent with the FISH-NanoSIMS observation that ANME-associated DSS were anabolically dormant without methane (Figure 2), as well as numerous previous reports showing SEEP-SRB1 in association with ANME-2 (Boetius et al., 2000; Orphan et al., 2001a; Pernthaler et al., 2008; Schreiber et al., 2010; Dekas et al., 2014).
Seep-SRB3 have not been observed in direct association with ANME. Members of a sister group to Seep-SRB3 associate with ANME-3 (Lösekann et al., 2007), but the Seep-SRB3 clones demonstrating methane-enhanced transcription here are distinct from the ANME-3 partner (Supplementary Figure 4). Additionally, mcrA transcripts affiliated with ANME-3 were not detected in these incubations (Figures 1a and b). The observation of enhanced rRNA expression in the presence of methane, although currently uncorroborated by additional data sets, suggest that members of Seep-SRB3 may benefit from methane and/or ANME-2 activity and warrants further investigation.
Are single cells of DSS and DSB active, comprising a truly ‘free-living’ population?
To differentiate activity between ANME-associated and single Deltaproteobacteria, we analyzed 15NH4+ uptake in individual cells with NanoSIMS. We used CARD-FISH with the oligonucleotide probe DSS_658 to target members of DSS (which includes the SEEP-SRB1 group; Knittel et al., 2003; Schreiber et al., 2010), and probe seepDBB_653 to target a broad group within the DSB, including members of SEEP-DBB and SEEP-SRB3/Desulfobulbus (Supplementary Table 4). We detect the uptake of 15NH4+ in individual DSS and DSB cells in the absence of CH4 (DSS: n=7 cells, 7/7 15N-enriched; DSB: n=7 cells, 4/7 15N-enriched; CR18). This indicates that both single-cell populations targeted by our FISH probes contain members that are anabolically active and not dependent on either CH4 or ANME activity (Figures 2c and 3b, c).
Additionally, compared with the lack of 15NH4+ incorporation in ANME-associated DSS in the same incubation (Figures 2c and 3a), the 15NH4+ incorporation measured in all single DSS cells suggests potentially fundamental physiological and likely phylogenetic differences between ANME-associated and physically independent DSS cells. In support of this, the aprA transcripts related to Desulfobacteraceae and Desulfobulbaceae recovered from the CH4 and Ar treatments were largely associated with different clades (Supplementary Figure 3).
Are diverse diazotrophs present and active in Mound 12 sediment?
A diversity of nifH sequences was detected in DNA extracted from the CH4 incubation (CR15), consistent with nifH diversity observed in hotspots of productivity in the deep sea, including methane seeps, mud volcanoes and hydrothermal vents (Mehta et al., 2003; Dang et al., 2009; Dekas et al., 2009; Miyazaki et al., 2009) (Figures 1a, b and 5). A nifH DNA clone library was previously generated from the same sediment sample immediately upon collection (not incubated) (Dekas et al., 2014), and the nifH diversity and proportions recovered in that library and the library reported here (after 4 months of incubation with CH4) are highly similar (Figure 1a). The similarity suggests that the experimental incubation contains a diazotrophic assemblage representative of the in situ population.

Translated nifH gene tree inferred with Maximum Likelihood using PhyML 3.0. Sequences from this study are bold. DNA clones are black and cDNA clones are red. For brevity, only a representative subset of the clones from this study is included in the tree. The total number of clones from each library that fall within the group of the clone shown (defined by the last well-supported branch, and indicated by the black square) is included in parentheses. SH-like aLRT (approximate Likelihood Ratio Test) branch supports above 0.70 for major branches are shown. The scale bar indicates the average number of amino acid substitutions per site. The tree was rooted with chlorophyllide reductase gene sequences YP_004716603, YP_680532, YP_001534851, and YP_508121. NCBI accession numbers are shown when possible; the IMG Gene IDs are listed for ANME-2D, ANME-2 A, and Desulfosarcina variabilis. nifH groups as described by Raymond et al., 2004 are indicated.
We detected nifH transcripts in the CH4 incubation, but not in the Ar incubation, consistent with previous work demonstrating methane-dependent nitrogen fixation at Mound 12 (Dekas et al., 2014) (Figure 1). This suggests that the diazotrophs are either dependent on methane (e.g., methanotrophs) or dependent on the products of the ANME (e.g., ANME-associated Deltaproteobacteria). ANME-2 fix nitrogen in methane seep sediment (Dekas et al., 2009), but other seep diazotrophs have not yet been identified. The methane seep nifH clade (‘methane seep group 2’ in Miyazaki et al., 2009) has been putatively assigned to the ANME-2 archaea (Dekas et al., 2009; Miyazaki et al., 2009), and our analysis supports this assignment with the placement of a nifH sequence recovered from an ANME-2a genome within this clade (Wang et al., 2014) (Figure 5). Interestingly, only 41% of the clones recovered in this study fall within the ANME-2-affiliated methane seep clade, whereas 49% fall within group III (49%) (Figure 5). Group III contains nifH sequences from a range of anaerobic microbes, including methanogens and Deltaproteobacterial sulfate-reducing bacteria. The transcription of these sequences suggests multiple methane-dependent diazotrophs within the sediments, potentially including ANME phylotypes in addition to ANME-2, or SRB with ANME-coupled activity.
Nitrogen fixation by ANME-associated Deltaproteobacteria is one possibility. DSS and DSB associated with ANME demonstrate 15N enrichment in the presence of 15N2 and methane (Dekas et al., 2009, 2014). Although this may be because of N sharing with the diazotrophic ANME-2, additional nitrogen fixation by the associated Deltaproteobacteria—although at lower rates—is also possible. Consistent with this, the nif genes recovered from nearly purified ANME-2c consortia in previous work were affiliated with both Methanosarcina-like and deltaproteobacterial nif sequences (Pernthaler et al., 2008). Why closely associated partners would both fix nitrogen is unclear, and to our knowledge, unprecedented. It raises the possibility that nitrogen fixation in these organisms is not because of N limitation, which could be overcome by an N2-fixing symbiont, but rather for other benefits only realized by the diazotroph itself. Further analyses, including metatranscriptomics/metaproteomics of individual ANME-Deltaproteobacteria consortia and/or immunolabeling of nitrogenase would be necessary to differentiate between the possibilities of N sharing between the diazotrophic ANME and associated Deltaproteobacteria, versus independent nitrogen fixation in both.
Are ANME-1, single DSS and single DSB capable of N2 fixation?
To investigate whether the diversity of nifH transcripts could be because of a diversity of diazotrophs in the single-cell population, we investigated 15N2 assimilation in single ANME-1, DSS and DSB cells using FISH-NanoSIMS. ANME-1 contain nif homologs, and although they are inferred to be non-functional in nitrogen fixation based on their phylogenetic placement within nifH group IV (Meyerdierks et al., 2010), we directly tested their ability to fix nitrogen in this study. Sediment from the ERB previously shown to host diazotrophic ANME-2 was chosen for this experiment rather than Mound 12 sediment because the incubated ERB sediment contained more ANME-1 cells (incubation ERB1C) (Dekas et al., 2009). NanoSIMS analysis revealed that single cells of ANME-1 did not fix nitrogen after a 6-month incubation with methane and 15N2 (n=10; Figure 3g and Supplementary Figure 5A). However, when we investigated the general anabolic activity of ANME-1 cells in parallel incubations with 15NH4+ and CH4, no 15N uptake was observed, suggesting these archaea were not active in the microcosm incubation (n=5; Supplementary Figure 5B). Conclusions regarding the ability of the ANME-1 to fix nitrogen therefore cannot be drawn. Consistent with previous studies (Nauhaus et al., 2005), the lack of NH4+ assimilation by ANME-1 in the same incubations where ANME-2 readily assimilated NH4+ (Dekas et al., 2009) suggests differences in the ecological physiology and optimal growth conditions for members of the ANME-1 and ANME-2.
Single sulfate-reducing bacteria are other likely candidates for N2 fixation in methane seep sediment, given their diazotrophic activity in shallow marine sediments (Bertics et al., 2010, 2012). Indeed, the majority of the nifH transcripts recovered in this study fall within a phylogenetic clade that includes nifH sequences affiliated with deltaproteobacterial sulfate-reducing bacteria (Figure 5). Single DSS and DSB cells were therefore targeted with FISH-NanoSIMS to determine whether or not they fix N2. However, after incubation with CH4 and 15N2, single DSS cells were not enriched in 15N (n=9; CR8). The lack of 15N2 incorporation observed for the single DSS does not eliminate the possibility of N2 fixation, but it suggests that if occurring, it is rare.
Interestingly, 33% of the single DSB cells analyzed did show 15N enrichment (n=21; CR8) (Figures 2a and 3d–f). Additional DAPI-stained microorganisms from the same incubation showed no 15N enrichment, indicating that recycling of 15N-enriched substrates was not significant over the course of the 5-month incubation (n=34; CR8) (Figures 2a and 3d). Without additional information, the 15N enrichment observed specifically in single DSB cells after incubation with 15N2 and methane would suggest diazotrophy. However, when FISH-NanoSIMS analyses were performed on single DSB cells in paired 15N2 incubations without CH4, surprisingly no 15N enrichment was observed (n=12, CR17; n=10, CR45) (Figure 2b). N2 fixation dependent on CH4 is unexpected for DSB, especially given that 15NH4+ uptake by the DSB was not CH4 dependent (Figure 2c).
Two scenarios could explain the pattern of 15N observed in the DSB single cells. The first is that free-living members of the DSB—although not the whole population—are both methane-dependent and diazotrophic. Although methane dependence in free-living SRB is unprecedented, some single DSB did not assimilate 15NH4+ without methane, leaving open the possibility of a methane-dependent sub-population. The second, and perhaps more parsimonious, is that single DSB cells are 15N-enriched because of a previous association with active ANME. In a previous study, DSB cells in association with ANME-2 were shown to be 15N-enriched after incubation with 15N2 and CH4 (Dekas et al., 2014). This could be due either to direct consumption of 15N2 (diazotrophy) when in association with ANME-2 or passage of fixed 15N products from diazotrophic ANME-2. If followed by disassociation from ANME, both scenarios would result in 15N-enriched single DSB cells only in the presence of methane, as we observed here. However, in the latter possibility, the DSB are not diazotrophic. Because of this, the combination of observations does not definitively provide evidence for DSB diazotrophy.
The data may, however, suggest an interesting difference between the ANME-2-DSS and ANME-2-DSB associations. The potential ANME-2-DSB dissociation could either be part of the life cycle of the ANME-2-DSB symbiosis or it could be an indication of a fragile association disrupted by the sampling procedure. Either way, this transient and/or fragile association is different from that observed for the DSS, which although always were 15N-enriched when associated with 15N-enriched ANME (Dekas et al., 2014) were never observed as 15N-enriched single cells in diazotrophic conditions (Figure 2a). The stability of the ANME-DSS connection was also observed in the incubation with argon and 15NH4+: although ANME-associated DSS cells were not 15N-enriched, all single DSS analyzed in the same incubation were. The ANME-2-DSS association may therefore either be more stable or physically stronger, compared with that of ANME-2-DSB.
Conclusions
The results yielded by FISH-NanoSIMS and transcript analysis of sediments from Mound 12 Costa Rica were consistent in some but not all cases (summarized in Supplementary Table 5). The inconsistencies likely resulted from the limitations of each method: transcripts do not definitely indicate activity and lack spatial information, and FISH-NanoSIMS measures activity (or lack thereof) in only a subset of the population. However, in combination, we were able to use these techniques to demonstrate that (1) single DSS and DSB cells are active within methane seep sediment, and are not dependent on methane and/or ANME activity, and (2) single DSS and ANME-associated DSS exhibit physiological differences with respect to their response to methane, suggesting phylogenetic differences. Additionally, we made observations suggesting that (1) a diversity of methane-dependent diazotrophs are active in Mound 12 sediment and may include ANME-associated Deltaproteobacteria, (2) the ANME-DSS association is stable, and may be more so than the ANME–DSB association, (3) ANME-1 and ANME-2 demonstrate differing growth rates and/or acceptable growth conditions and (4) Seep-SRB1, and more surprisingly, Seep-SRB3 show a positive transcriptional response to methane/ANME activity. Taken together, these insights provide new understanding of the dynamics between ANME and seep Deltaproteobacteria, and highlight the complimentary nature of transcript and FISH-NanoSIMS analyses to assess microbial activity.
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
Alperin M, Hoehler T . (2009). Anaerobic methane oxidation by archaea/sulfate-reducing bacteria aggregates: 1. Thermodynamic and physical constraints. Am J Sci 309: 869–957.
Anisimova M, Gascuel O . (2006). Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol 55: 539–552.
Bertics VJ, Loscher CR, Salonen I, Dale AW, Schmitz RA, Treude T . (2012). Occurrence of benthic microbial nitrogen fixation coupled to sulfate reduction in the seasonally hypoxic Eckernforde Bay, Baltic Sea. Biogeosci Discuss 9: 6489–6533.
Bertics VJ, Sohm JA, Treude T, Chow CET, Capone DG, Fuhrman JA et al. (2010). Burrowing deeper into benthic nitrogen cycling: the impact of bioturbation on nitrogen fixation coupled to sulfate reduction. Mar Ecol Prog Ser 409: 1–15.
Blazewicz SJ, Barnard RL, Daly RA, Firestone MK . (2013). Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J 7: 2061–2068.
Boetius A, Ravenschlag K, Schubert CJ, Rickert D, Widdel F, Gieseke A et al. (2000). A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407: 623–626.
Dang H, Luan X, Zhao J, Li J . (2009). Diverse and novel nifH and nifH-like gene sequences in the deep-sea methane seep sediments of the Okhotsk Sea. Appl Environ Microbiol 75: 2238–2245.
Dekas AE, Chadwick GL, Bowles MW, Joye SB, Orphan VJ . (2014). Spatial distribution of nitrogen fixation in methane seep sediment and the role of the ANME archaea. Environ Microbiol 16: 3012–3029.
Dekas AE, Orphan VJ . (2011). Identification of diazotrophic microorganisms in marine sediment via fluorescence in situ hybridization coupled to nanoscale secondary ion mass spectrometry (FISH-Nanosims). Methods Enzymol 486: 281–305.
Dekas AE, Poretsky RS, Orphan VJ . (2009). Deep-sea archaea fix and share nitrogen in methane-consuming microbial consortia. Science 326: 422–426.
Fordyce SL, Kampmann ML, van Doorn NL, Gilbert MT . (2013). Long-term RNA persistence in postmortem contexts. Invest Genet 4: 7.
Fulweiler RW, Brown SM, Nixon SW, Jenkins BD . (2013). Evidence and a conceptual model for the co-occurrence of nitrogen fixation and denitrification in heterotrophic marine sediments. Mar Ecol Prog Ser 482: 57–68.
Girguis PR, Cozen AE, DeLong EF . (2005). Growth and population dynamics of anaerobic methane-oxidizing archaea and sulfate-reducing bacteria in a continuous flow bioreactor. Appl Environ Microbiol 71: 3725–3733.
Green-Saxena AM, Dekas AE, Dalleska NF, Orphan VJ . (2014). Nitrate-based niche differentiation by distinct sulfate-reducing bacteria involved in the anaerobic oxidation of methane. ISME J 8: 150–163.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O . (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59: 307–321.
Hallam SJ, Girguis PR, Preston CM, Richardson PM, DeLong EF . (2003). Identification of methyl coenzyme M reductase A (mcrA) genes associated with methane-oxidizing archaea. Appl Environ Microbiol 69: 5483–5491.
Hallam SJ, Putnam N, Preston CM, Detter JC, Rokhsar D, Richardson PM et al. (2004). Reverse methanogenesis: testing the hypothesis with environmental genomics. Science 305: 1457–1462.
Hoehler T, Alperin M, Albert D, Martens C . (1994). Field and laboratory studies of methane oxidation in an anoxic marine sediment: evidence for a methanogen-sulfate reducer consortium. Global Biogeochem Cy 8: 451–463.
Holler T, Widdel F, Knittel K, Amann R, Kellermann MY, Hinrichs KU et al. (2011). Thermophilic anaerobic oxidation of methane by marine microbial consortia. ISME J 5: 1946–1956.
House CH, Orphan VJ, Turk KA, Thomas B, Pernthaler A, Vrentas JM et al. (2009). Extensive carbon isotopic heterogeneity among methane seep microbiota. Environ Microbiol 11: 2207–2215.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649.
Kessler PS, Leigh JA . (1999). Genetics of nitrogen regulation in Methanococcus maripaludis. Genetics 152: 1343–1351.
Kleindienst S, Ramette A, Amann R, Knittel K . (2012). Distribution and in situ abundance of sulfate-reducing bacteria in diverse marine hydrocarbon seep sediments. Environ Microbiol 14: 2689–2710.
Knittel K, Boetius A, Lemke A, Eilers H, Lochte K, Pfannkuche O et al. (2003). Activity, distribution, and diversity of sulfate reducers and other bacteria in sediments above gas hydrate (Cascadia margin, Oregon). Geomicrobiol J 20: 269–294.
Krueger M, Wolters H, Gehre M, Joye SB, Richnow HH . (2008). Tracing the slow growth of anaerobic methane-oxidizing communities by N-15-labelling techniques. FEMS Microbiol Ecol 63: 401–411.
Lane DJ . (1991) 16 S/23 S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds). Nucleic Acid Techniques in Bacterial Systematics. Wiley: : New York, NY, USA, pp 115–147.
Le SQ, Gascuel O . (2008). An improved general amino acid replacement matrix. Mol Biol Evol 25: 1307–1320.
Liang JH, Nielsen GM, Lies DP, Burris RH, Roberts GP, Ludden PW . (1991). Mutations in the draT and draG genes of Rhodospirillum rubrum result in loss of regulation of nitrogenase by reversible ADP-ribosylation. J Bacteriol 173: 6903–6909.
Lloyd KG, Alperin MJ, Teske A . (2011). Environmental evidence for net methane production and oxidation in putative ANaerobic MEthanotrophic (ANME) archaea. Environ Microbiol 13: 2548–2564.
Lösekann T, Knittel K, Nadalig T, Fuchs B, Niemann H, Boetius A et al. (2007). Diversity and abundance of aerobic and anaerobic methane oxidizers at the Haakon Mosby mud volcano, Barents Sea. Appl Environ Microbiol 73: 3348–3362.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
Luton PE, Wayne JM, Sharp RJ, Riley PW . (2002). The mcrA gene as an alternative to 16 S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology 148: 3521–3530.
Mehta MP, Butterfield DA, Baross JA . (2003). Phylogenetic diversity of nitrogenase (nifH) genes in deep-sea and hydrothermal vent environments of the Juan de Fuca Ridge. Appl Environ Microbiol 69: 960–970.
Meyer B, Kuever J . (2007). Molecular analysis of the diversity of sulfate-reducing and sulfur-oxidizing prokaryotes in the environment, using aprA as functional marker gene. Appl Environ Microbiol 73: 7664–7679.
Meyerdierks A, Kube M, Kostadinov I, Teeling H, Glockner FO, Reinhardt R et al. (2010). Metagenome and mRNA expression analyses of anaerobic methanotrophic archaea of the ANME-1 group. Environ Microbiol 12: 422–439.
Milucka J, Ferdelman TG, Polerecky L, Franzke D, Wegener G, Schmid M et al. (2012). Zero-valent sulphur is a key intermediate in marine methane oxidation. Nature 491: 541–546.
Miyazaki J, Higa R, Toki T, Ashi J, Tsunogai U, Nunoura T et al. (2009). Molecular characterization of potential nitrogen fixation by anaerobic methane-oxidizing archaea in the methane seep sediments at the number 8 Kumano Knoll in the Kumano Basin, offshore of Japan. Appl Environ Microbiol 75: 7153–7162.
Moran JJ, Beal EJ, Vrentas JM, Orphan VJ, Freeman KH, House CH . (2008). Methyl sulfides as intermediates in the anaerobic oxidation of methane. Environ Microbiol 10: 162–173.
Nauhaus K, Albrecht M, Elvert M, Boetius A, Widdel F . (2007). In vitro cell growth of marine archaealbacterial consortia during anaerobic oxidation of methane with sulfate. Environ Microbiol 9: 187–196.
Nauhaus K, Boetius A, Kruger M, Widdel F . (2002). In vitro demonstration of anaerobic oxidation of methane coupled to sulphate reduction in sediment from a marine gas hydrate area. Environ Microbiol 4: 296–305.
Nauhaus K, Treude T, Boetius A, Kruger M . (2005). Environmental regulation of the anaerobic oxidation of methane: a comparison of ANME-I and ANME-II communities. Environ Microbiol 7: 98–106.
Niemann H, Losekann T, de Beer D, Elvert M, Nadalig T, Knittel K et al. (2006). Novel microbial communities of the Haakon Mosby mud volcano and their role as a methane sink. Nature 443: 854–858.
Orcutt B, Boetius A, Elvert M, Samarkin V, Joye SB . (2005). Molecular biogeochemistry of sulfate reduction, methanogenesis and the anaerobic oxidation of methane at Gulf of Mexico cold seeps. Geochim Cosmochim Acta 69: 4267–4281.
Orcutt B, Samarkin V, Boetius A, Joye S . (2008). On the relationship between methane production and oxidation by anaerobic methanotrophic communities from cold seeps of the Gulf of Mexico. Environ Microbiol 10: 1108–1117.
Orphan VJ, Hinrichs KU, Ussler W III, Paull CK, Taylor LT, Sylva SP et al. (2001a). Comparative analysis of methane-oxidizing archaea and sulfate-reducing bacteria in anoxic marine sediments. Appl Environ Microbiol 67: 1922–1934.
Orphan VJ, House CH, Hinrichs KU, McKeegan KD, DeLong EF . (2001b). Methane-consuming archaea revealed by directly coupled isotopic and phylogenetic analysis. Science 293: 484–487.
Orphan VJ, House CH, Hinrichs KU, McKeegan KD, DeLong EF . (2002). Multiple archaeal groups mediate methane oxidation in anoxic cold seep sediments. Proc Natl Acad Sci USA 99: 7663–7668.
Orphan VJ, Turk KA, Green AM, House CH . (2009). Patterns of 15N assimilation and growth of met1hanotrophic ANME-2 archaea and sulfate-reducing bacteria within structured syntrophic consortia revealed by FISH-SIMS. Environ Microbiol 11: 1777–1791.
Pernthaler A, Pernthaler J . (2007). Fluorescence in situ hybridization for the identification of environmental microbes. Methods Mol Biol 353: 153–164.
Pernthaler A, Dekas AE, Brown CT, Goffredi SK, Embaye T, Orphan VJ . (2008). Diverse syntrophic partnerships from deep-sea methane vents revealed by direct cell capture and metagenomics. Proc Natl Acad Sci USA 105: 7052–7057.
Pernthaler A, Pernthaler J, Amann R . (2002). Fluorescence in situ hybridization and catalyzed reporter deposition for the identification of marine bacteria. Appl Environ Microbiol 68: 3094–3101.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41: D590–D596.
Raymond J, Siefert JL, Staples CR, Blankenship RE . (2004). The natural history of nitrogen fixation. Mol Biol Evol 21: 541–554.
Reeburgh WS . (2007). Oceanic methane biogeochemistry. Chem Rev 107: 486–513.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61: 539–542.
Schreiber L, Holler T, Knittel K, Meyerdierks A, Amann R . (2010). Identification of the dominant sulfate-reducing bacterial partner of anaerobic methanotrophs of the ANME-2 clade. Environ Microbiol 12: 2327–2340.
Sukenik A, Kaplan-Levy RN, Welch JM, Post AF . (2012). Massive multiplication of genome and ribosomes in dormant cells (akinetes) of Aphanizomenon ovalisporum (Cyanobacteria). ISME J 6: 670–679.
Vigneron A, Cruaud P, Pignet P, Caprais JC, Gayet N, Cambon-Bonavita MA et al. (2014). Bacterial communities and syntrophic associations involved in anaerobic oxidation of methane process of the Sonora Margin cold seeps, Guaymas Basin. Environ Microbiol 16: 2777–2790.
Waldbauer JR, Rodrigue S, Coleman ML, Chisholm SW . (2012). Transcriptome and proteome dynamics of a light-dark synchronized bacterial cell cycle. PLoS One 7: e43432.
Wang FP, Zhang Y, Chen Y, He Y, Qi J, Hinrichs KU et al. (2014). Methanotrophic archaea possessing diverging methane-oxidizing and electron-transporting pathways. ISME J 8: 1069–1078.
Zhang Y, Burris RH, Ludden PW, Roberts GP . (1993). Posttranslational regulation of nitrogenase activity by anaerobiosis and ammonium in Azospirillum brasilense. J Bacteriol 175: 6781–6788.
Acknowledgements
We thank the pilots, crew and science party of AT15-11 and AT15-59, especially Lisa Levin, Jake Bailey and Shana Goffredi; Abigail Green-Saxena and Joshua Steele for valuable discussions; Bethany Jenkins for consultation with RNA extraction protocol development; John Eiler and Yunbin Guan for assistance with NanoSIMS measurements. We additionally thank three anonymous reviewers for their careful critique of the manuscript. Funding was provided by the US Department of Energy, Office of Science, Office of Biological and Environmental Research (DE-SC0003940 and DE-SC0004949 to VJO), the National Science Foundation (MCB-0348492 to VJO and a Graduate Research Fellowship to AED) and the Gordon and Betty Moore Foundation (GBMF no. 3780 to VJO, and via support for The Caltech Center for Microanalysis housing the CAMECA NanoSIMS 50 L). The writing of this manuscript by AED was partially performed while funded by a Lawrence Postdoctoral Fellowship under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dekas, A., Connon, S., Chadwick, G. et al. Activity and interactions of methane seep microorganisms assessed by parallel transcription and FISH-NanoSIMS analyses. ISME J 10, 678–692 (2016). https://doi.org/10.1038/ismej.2015.145
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.145
This article is cited by
-
Single-cell view of deep-sea microbial activity and intracommunity heterogeneity
The ISME Journal (2023)
-
Phylogenetically and catabolically diverse diazotrophs reside in deep-sea cold seep sediments
Nature Communications (2022)
-
The use of stable carbon isotopes to decipher global change effects on soil organic carbon: present status, limitations, and future prospects
Biogeochemistry (2022)
-
Evidence for phylogenetically and catabolically diverse active diazotrophs in deep-sea sediment
The ISME Journal (2020)
